- High School
- You don't have any recent items yet.
- You don't have any courses yet.
- You don't have any books yet.
- You don't have any Studylists yet.
- Information

Drug metabolism essay - Grade: 88
Pharmacology and health (sw 246), depaul university, students also viewed.
- Hyperlipidemia and Atherosclerosis
- Congestive Heart Failure
- Hypertension
- Autonomic Pharmacology
- Pharmacology and Health chpt 12
- Pharmacology and Health chpt 11
Related documents
- Pharmacology and Health Chpt 10
- Pharmacology and Health Chpt 9
- Pharmacology and Health Chpt 8
- Pharmacology and Health Chpt7
- Pharmacology and Health Chpt 6
- Pharmacology and Health Chpt 5
Course : Pharmacology And Health (SW 246)
University : depaul university.

- More from: Pharmacology And Health SW 246 DePaul University 17 Documents Go to course

An official website of the United States government
The .gov means it's official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- Browse Titles
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.

StatPearls [Internet].
Drug metabolism.
Stephen T. Susa ; Azhar Hussain ; Charles V. Preuss .
Affiliations
Last Update: August 17, 2023 .
- Continuing Education Activity
Most drugs undergo chemical alteration by various bodily systems to create compounds that are more easily excreted from the body. These chemical alterations occur primarily in the liver and are known as biotransformations. Understanding the chemical alterations drugs undergo as they are metabolized is relevant when planning individual pharmacological interventions for patients. This activity reviews drug metabolism, biotransformations, and polypharmacy. The role of the interprofessional team in caring for patients using multiple medications is discussed.
- Differentiate between phase I (oxidation, reduction, hydrolysis) and phase II (conjugation) reactions.
- Assess the impact of drug metabolism on treatment response and drug interactions, considering the potential for drug toxicity or therapeutic failure.
- Screen patients for factors affecting drug metabolism, such as genetic polymorphisms, drug-drug interactions, and organ dysfunction.
- Coordinate with healthcare teams to ensure the integration of drug metabolism information into treatment plans, promoting personalized and evidence-based care.
- Introduction
Most drugs are xenobiotics, ie, chemical substances not naturally produced by the body. Xenobiotics undergo various body processes for detoxification, thus reducing their toxicity and allowing them to be readily available for excretion. These processes allow for the chemical modification of drugs into their metabolites and are known as drug metabolism or metabolic biotransformation. [1] [2]
These metabolites are the byproducts of drug metabolism and can be characterized by active, inactive, and toxic metabolites. Active metabolites are biochemically active compounds with therapeutic effects, whereas inactive metabolites are biochemically inactive compounds with neither a therapeutic nor toxic effect. Toxic metabolites are biochemically active compounds similar to active metabolites but have various harmful effects. [3]
Drug metabolism occurs at a specific location in the body, resulting in a low concentration of active metabolites in the systemic circulation. This phenomenon is called first-pass metabolism because it limits drug bioavailability. First-pass metabolism primarily occurs in the liver; however, metabolizing enzymes can be found throughout the body. [2] [3]
Understanding these alterations in chemical activity is crucial in utilizing the optimal pharmacological intervention for any patient. This is a topic of interest to any provider who routinely treats patients with medications. [1] [2] [3] [4]
The kidneys are primarily responsible for the excretion of drugs from the body; however, lipophilic drugs readily cross the cell membrane of the kidney tubules and are reabsorbed into the blood. [3] Therefore, lipophilic drugs are first metabolized in the liver before excretion of the drug can be possible. The metabolism of drugs can occur in various reactions, categorized as phase I (modification), phase II (conjugation), and in some instances, phase III (additional modification and excretion).
Phase I modifications alter the lipophilic drug chemical structure through oxidation, reduction, hydrolysis, cyclization/decyclization, and either by removing hydrogen or adding oxygen to more polar molecules. In some instances, this process changes an inactive prodrug into a metabolically active drug. Oxidation typically results in metabolites that still retain some of their pharmacological activity. For example, phase I modification transforms the common anxiolytic drug diazepam into desmethyldiazepam and then further into oxazepam. Both of those metabolites produce similar physiological and psychological effects to diazepam itself. The cytochrome P450 system, also known as microsomal mixed function oxidase, catalyzes most phase I reactions. [3] [5]
In phase II modifications, a drug molecule couples with another molecule in a conjugation reaction. Conjugation usually renders the compound pharmacologically inert and water-soluble, allowing the compound to be easily excreted. Conjugation mechanisms include methylation, acetylation, sulphation, glucuronidation, and glycine or glutathione conjugation. These processes can occur in various locations, such as the liver, kidney, lungs, intestines, and other organ systems. An example of phase II metabolism is oxazepam, which conjugates with another molecule called glucuronide. The drug becomes physiologically inactive and is excreted without further chemical modification. [5] [6]
Phase III metabolism may also follow phase II metabolism, in which conjugates and metabolites are excreted from the cells. A critical factor in drug metabolism is the enzymatic catalysis of phase I and II processes. The type and concentration of liver enzymes are crucial to the efficient metabolism of drugs. The most commonly used enzymes for medical purposes are monoamine oxidase and cytochrome P450. These 2 enzymes are responsible for metabolizing dozens of biogenic and xenobiotic chemicals.
As the name suggests, monoamine oxidase catalyzes the processing of monoamines such as serotonin and dopamine. Monoamine oxidase inhibitors (MAOI) are used as antidepressants that increase serotonin and dopamine levels in the brain. The cytochrome P450 system is a family of heme-containing isoenzymes, primarily located in the liver and gastrointestinal tract, responsible for metabolizing many drugs and compounds, such as lipids and steroids. [7] Cytochrome P450 catalyzes the metabolism of many psychoactive drugs, including amphetamines and opioids. [8]
- Issues of Concern
Drug metabolism can affect the plasma concentrations of drugs, which must be considered. Prescribers must be concerned about drug-drug interactions, as they may negatively impact the patient's health. For example, if rifampin is taken concurrently with imatinib, imatinib's plasma concentrations can be reduced because rifampin can induce CYP3A4 activity and metabolize imatinib at a much faster rate. Thus, imatinib's anticancer activity can be attenuated. [7] [8]
Similarly, if the patient takes omeprazole to treat gastroesophageal reflux disease (GERD) concurrently with imatinib, imatinib's plasma concentrations increase. This occurs because omeprazole is a potent inhibitor of CYP3A4, and CYP3A4 metabolizes imatinib. The inhibition of CYP3A4 causes the accumulation of imatinib, leading to toxicities. [7] [8] [9]
Aging is another factor that affects drug metabolism. Drug metabolism slows with age, making older patients more susceptible to adverse reactions. Prescribers should keep in mind the patient's age when prescribing medication. Genetic polymorphism of drug-metabolizing enzymes can also cause variations in drug effects, leading to patients reacting differently to various drugs. [9]
- Clinical Significance
In any pharmacological intervention, the prescriber must consider how and when a specific drug is eliminated from the body. Most of the time, drug clearance occurs according to first-order kinetics; in other words, the clearance rate depends on the drug's plasma concentration. That is, the elimination rate is proportional to the drug's concentration. The rate of this form of clearance depends upon the chemical in question and is often represented by a half-life. This is the time it takes for 50% of the drug to be eliminated from the body. For example, the half-life of cocaine is approximately 1 hour; thus, after 4 hours, only about 6.25% of the initial dose is present within the body. [10]
However, the elimination of some drugs occurs at a constant rate that is independent of plasma concentrations. Ethanol is an example; it is eliminated at a constant rate of about 15 mL/hour regardless of the concentration in the bloodstream. This is called zero-order kinetics and occurs when enzyme binding sites are saturated at low concentrations. Kinetics are of interest in medicine because monitoring drug concentration is often of clinical importance with many medications. Understanding pharmacokinetics, specifically drug elimination, allows providers to alter patient-specific therapies. Therapy aims to achieve a steady-state plasma concentration at which drug metabolism and elimination occur simultaneously. [10] [11]
Metabolism is a highly variable process that can be influenced by several factors. One major disruptor of drug metabolism is depot binding, meaning the coupling of drug molecules to inactive sites in the body such that the drug is no longer accessible for metabolism. This can affect the duration of action of pharmacological agents susceptible to depot binding. One notable example is tetrahydrocannabinol (THC), the main psychoactive component of marijuana. THC is highly lipid-soluble, and depot binds in the adipose tissue of users. This interaction drastically slows the drug's metabolism, which is why metabolites of THC can be detected in urine weeks after the patient's last use. [10] [11] [12] [13]
Another factor in drug metabolism is enzyme induction. Enzymes are induced by repeated use of the same chemical. The body becomes accustomed to the constant presence of the drug and compensates by increasing the production of enzymes necessary for the drug's metabolism. This contributes to pharmacological tolerance and is one reason patients need ever-increasing doses of certain drugs to produce the same effect.
Opioids are a prime example. Patients with long-term prescriptions for opioid analgesics notice their medication becomes less effective over time. Notably, induction increases the metabolic rate for all drugs processed via the enzyme induced; for example, chronic amphetamine use causes higher concentrations of CYP2D6. This enzyme is also essential in the metabolism of certain opioids, such as oxycodone; thus, a physician prescribing oxycodone to a patient using amphetamines would have to prescribe a higher dose to produce the desired effect. [11]
In contrast, some drugs have an inhibitory effect on enzymes, making the patient more sensitive to other medications metabolized through the action of those enzymes. A classic example is the inhibition of monoamine oxidase by certain antidepressant drugs. These compounds produce psychotherapeutic effects by blocking the enzyme that breaks down 'pleasure' chemicals in the brain. However, this can cause problems when patients on an MAOI take other drugs, which can cause abnormally high concentrations of these neurochemicals. A patient on an MAOI who uses cocaine, which elevates the concentration of serotonin, dopamine, and norepinephrine, will experience a much more potent effect from the cocaine. This interaction can lead to numerous physiological problems, including tachycardia, hypertension, and serotonin syndrome. [12] [14]
Drugs that share elements of their metabolic pathways can also 'compete' for the same binding sites on enzymes, decreasing their metabolism's efficiency. For instance, alcohol and certain sedatives are metabolized by the same member of the cytochrome P450 family. Only a limited number of enzymes exist to break these chemicals down. Thus, if pentobarbital is administered to a patient who is also metabolizing alcohol, the pentobarbital would not be completely metabolized because most of the necessary enzymes would be filled by alcohol molecules; this is one reason that alcohol and other sedative or hypnotic drugs can have a synergistic effect when coadministered. [7] [8]
- Other Issues
Regardless of whether a drug is renally or hepatically metabolized and eliminated, impairment in either of these systems can cause significant issues. These issues can include altered dosing, dose intervals, and therapeutic effect; the pathway for metabolism can determine if a particular drug can be prescribed and used for a specific patient. [15]
- Enhancing Healthcare Team Outcomes
Drug metabolism is an essential clinical concern for the interprofessional healthcare team. Clinicians and pharmacists must work together to prevent clinically significant drug interactions that could affect patients' health.
In a hospital setting, nursing staff monitors for signs of a toxic buildup of metabolites or active drugs. This is especially significant in renal or hepatic insufficiency. In many cases, drugs such as aminoglycoside antibiotics, warfarin, and fluoroquinolones are dosed and monitored by pharmacists, who monitor serum levels of the drugs and renal function.
An interprofessional approach to drug dosing and administration in light of the effects of drug metabolism on patients, whether through impaired metabolism, drug-drug interactions, enzymatic induction, or other factors, provides the best potential for optimal patient care. The interprofessional care approach results in better therapeutic results with fewer adverse events.
- Nursing, Allied Health, and Interprofessional Team Interventions
Interprofessional interventions and monitoring are critical for positive outcomes. Literature suggests that interprofessional interventions are effective when teams are open and willing to collaborate, communicate, share decision-making, and coordinate care, ultimately leading to the integration of the team’s competencies. [16]
Interprofessional team interventions are an effective part of medication management through interventions and monitoring. Medications affect people differently, as mentioned above. As a result, patients may be at an increased risk of adverse events if appropriate measures are not taken. Teams must monitor and intervene when medications are not safe for patients to take or administer or if patients are taking them inappropriately. The goal should always be patient safety. Reporting adverse events is crucial for interprofessional team monitoring. To properly conduct monitoring and interventions, clinical teams should also undergo training.
Training can be conducted on topics such as identifying medication errors to prevent adverse events, effective communication amongst teams, and standardization of medication dispensing. Continuing Medication Credits should be geared toward intervention and monitoring as well. Communication is a critical part of healthcare delivery. Interprofessional teams must continue to advance their approaches to following patients through the continuum of care.
In addition, effective interprofessional training allows for increased quality of care, promotion of collaboration, promote interdisciplinary awareness, respect, and acceptability of the role each discipline plays. However, one should also consider the challenges as well. Each discipline has its own culture and methodology. They have differences in routines, regulations, qualifications, accountability, and professional language. As a result, it makes it difficult to standardize any interventions or monitoring of medications. However, the first step in change is awareness of these challenges. Once these challenges are addressed and each discipline, whether nurses, pharmacists, or physicians, is trained appropriately, they will be able to effectively intervene and monitor medications for improved health outcomes at any stage of treatment. [17]
- Review Questions
- Access free multiple choice questions on this topic.
- Comment on this article.
Disclosure: Stephen Susa declares no relevant financial relationships with ineligible companies.
Disclosure: Azhar Hussain declares no relevant financial relationships with ineligible companies.
Disclosure: Charles Preuss declares no relevant financial relationships with ineligible companies.
This book is distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0) ( http://creativecommons.org/licenses/by-nc-nd/4.0/ ), which permits others to distribute the work, provided that the article is not altered or used commercially. You are not required to obtain permission to distribute this article, provided that you credit the author and journal.
- Cite this Page Susa ST, Hussain A, Preuss CV. Drug Metabolism. [Updated 2023 Aug 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.
In this Page
Bulk download.
- Bulk download StatPearls data from FTP
Related information
- PMC PubMed Central citations
- PubMed Links to PubMed
Similar articles in PubMed
- [Xenobiotic toxicity prediction combined with xenobiotic metabolism prediction in the human body]. [Biomed Khim. 2019] [Xenobiotic toxicity prediction combined with xenobiotic metabolism prediction in the human body]. Rudik AV, Dmitriev AV, Lagunin AA, Ivanov SM, Filimonov DA, Poroikov VV. Biomed Khim. 2019 Feb; 65(2):114-122.
- Review Drug metabolism for the perplexed medicinal chemist. [Chem Biodivers. 2009] Review Drug metabolism for the perplexed medicinal chemist. Testa B. Chem Biodivers. 2009 Nov; 6(11):2055-70.
- Review Drug-metabolizing enzymes: mechanisms and functions. [Curr Drug Metab. 2000] Review Drug-metabolizing enzymes: mechanisms and functions. Sheweita SA. Curr Drug Metab. 2000 Sep; 1(2):107-32.
- Review Biotransformation enzymes as determinants of xenobiotic toxicity in domestic animals. [Vet J. 2001] Review Biotransformation enzymes as determinants of xenobiotic toxicity in domestic animals. Nebbia C. Vet J. 2001 May; 161(3):238-52.
- Review Opportunities for Accelerating Drug Discovery and Development by Using Engineered Drug-Metabolizing Enzymes. [Drug Metab Dispos. 2023] Review Opportunities for Accelerating Drug Discovery and Development by Using Engineered Drug-Metabolizing Enzymes. Gillam EMJ, Kramlinger VM. Drug Metab Dispos. 2023 Mar; 51(3):392-402. Epub 2022 Dec 2.
Recent Activity
- Drug Metabolism - StatPearls Drug Metabolism - StatPearls
Your browsing activity is empty.
Activity recording is turned off.
Turn recording back on
Connect with NLM
National Library of Medicine 8600 Rockville Pike Bethesda, MD 20894
Web Policies FOIA HHS Vulnerability Disclosure
Help Accessibility Careers
Drug Metabolism
The liver is the principal site of drug metabolism (for review, see [ 1 ]). Although metabolism typically inactivates drugs, some drug metabolites are pharmacologically active—sometimes even more so than the parent compound. An inactive or weakly active substance that has an active metabolite is called a prodrug, especially if designed to deliver the active moiety more effectively.
Drugs can be metabolized by oxidation, reduction, hydrolysis, hydration, conjugation, condensation, or isomerization; whatever the process, the goal is to make the drug easier to excrete. The enzymes involved in metabolism are present in many tissues but generally are more concentrated in the liver.
Drug metabolism rates vary among patients. Some patients metabolize a drug so rapidly that therapeutically effective blood and tissue concentrations are not reached; in others, metabolism may be so slow that usual doses have toxic effects. Individual drug metabolism rates are influenced by genetic factors, coexisting disorders (particularly chronic liver disorders and advanced heart failure), and drug interactions (especially those involving induction or inhibition of metabolism).
For many drugs, metabolism occurs in 2 phases.
Phase I reactions involve formation of a new or modified functional group or cleavage (oxidation, reduction, hydrolysis); these reactions are nonsynthetic.
Phase II reactions involve conjugation with an endogenous substance (eg, glucuronic acid, sulfate, glycine ); these reactions are synthetic.
Metabolites formed in synthetic reactions are more polar and thus more readily excreted by the kidneys (in urine) and the liver (in bile) than those formed in nonsynthetic reactions. Some drugs undergo only phase I or phase II reactions; thus, phase numbers reflect functional rather than sequential classification.
Hepatic drug transporters are present throughout parenchymal liver cells and affect a drug’s liver disposition, metabolism, and elimination (for review, see [ 1 , 2 ]). The 2 primary types of transporters are
Influx, which translocate molecules into the liver
Efflux, which mediate excretion of drugs into the blood or bile
Genetic polymorphisms can variably affect the expression and function of hepatic drug transporters to potentially alter a patient's susceptibility to drug adverse effects and drug-induced liver injury . For example, carriers of certain transporter genotypes exhibit increased blood levels of statins and are more susceptible to statin-induced myopathy when statins are used for the treatment of hypercholesterolemia ( 1 , 2 ).
(See also Overview of Pharmacokinetics .)
For almost all drugs, the metabolism rate in any given pathway has an upper limit (capacity limitation). However, at therapeutic concentrations of most drugs, usually only a small fraction of the metabolizing enzyme’s sites are occupied, and the metabolism rate increases with drug concentration. In such cases, called first-order elimination (or kinetics), the metabolism rate of the drug is a constant fraction of the drug remaining in the body (ie, the drug has a specific half-life).
For example, if 500 mg is present in the body at time zero, after metabolism, 250 mg may be present at 1 hour and 125 mg at 2 hours (illustrating a half-life of 1 hour). However, when most of the enzyme sites are occupied, metabolism occurs at its maximal rate and does not change in proportion to drug concentration; instead, a fixed amount of drug is metabolized per unit time (zero-order kinetics). In this case, if 500 mg is present in the body at time zero, after metabolism, 450 mg may be present at 1 hour and 400 mg at 2 hours (illustrating a maximal clearance of 50 mg/hour and no specific half-life). As drug concentration increases, metabolism shifts from first-order to zero-order kinetics.
Cytochrome P-450
The most important enzyme system of phase I metabolism is cytochrome P-450 (CYP450), a microsomal superfamily of isoenzymes that catalyzes the oxidation of many drugs. The electrons are supplied by nicotinamide adenine dinucleotide phosphate (NADPH)–CYP450 reductase, a flavoprotein that transfers electrons from NADPH (the reduced form of nicotinamide adenine dinucleotide phosphate) to CYP450.
CYP450 enzymes can be induced or inhibited by many drugs and substances resulting in drug interactions in which one drug enhances the toxicity or reduces the therapeutic effect of another drug. For examples of drugs that interact with specific enzymes, see tables Common Substances That Interact With Cytochrome P-450 Enzymes and Drug Interactions .
Common Substances That Interact With Cytochrome P-450 Enzymes
With aging, the liver’s capacity for metabolism through the CYP450 enzyme system is reduced by ≥ 30% because hepatic volume and blood flow are decreased. Thus, drugs that are metabolized through this system reach higher levels and have prolonged half-lives in older adults (see figure Comparison of pharmacokinetic outcomes for diazepam in a younger man [A]... ). Because neonates have partially developed hepatic microsomal enzyme systems, they also have difficulty metabolizing many drugs.
Conjugation
Glucuronidation, the most common phase II reaction, is the only one that occurs in the liver microsomal enzyme system. Glucuronides are secreted in bile and eliminated in urine. Thus, conjugation makes most drugs more soluble and easily excreted by the kidneys. Amino acid conjugation with glutamine or glycine produces conjugates that are readily excreted in urine but not extensively secreted in bile. Aging does not affect glucuronidation. However, in neonates, conversion to glucuronide is slow, potentially resulting in serious effects (eg, as with chloramphenicol ).
Conjugation may also occur through acetylation or sulfoconjugation. Sulfate esters are polar and readily excreted in urine. Aging does not affect these processes.
1. Patel M, Taskar KS, Zamek-Gliszczynski MJ : Importance of hepatic transporters in clinical disposition of drugs and their metabolites. J Clin Pharmacol 56(Suppl 7):S23–S39, 2016. doi: 10.1002/jcph.671
2. Pan G : Roles of hepatic drug transporters in drug disposition and liver toxicity. Adv Exp Med Biol 1141:293-340, 2019. doi:10.1007/978-981-13-7647-4_6

Copyright © 2024 Merck & Co., Inc., Rahway, NJ, USA and its affiliates. All rights reserved.
- Cookie Preferences

Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
5 Drug Metabolism
“I can’t change the direction of the wind, but I can adjust my sails to always reach my destination.”
– Jimmy Dean
5.1 Biological Pathways for Drug Metabolism
Following administration, drugs are absorbed and distributed into different tissues. Over time, these molecules are eliminated (irreversibly removed) from the body which can occur in two ways: either directly unchanged (excretion/elimination) or changed (metabolized). Drug elimination predominantly occurs through the kidneys (especially for hydrophilic species) and exits via urine, although a subset of drugs (predominantly non-polar substances) are eliminated in the bile (via action of the liver) and excreted in the feces.
The liver is the principal site for drug processing and metabolism and has (without exaggeration) hundreds of characterized functions including roles in digestion, detoxification, immunity, hormone release, ammonia processing, glycogen production, and heavy metal storage. The liver also produces bile, which is a mixture comprised of sterol derivatives (bile salts), cholesterol, phospholipids, and electrolytes, that is stored in the gall bladder and released in the small intestine to facilitate digestion of fats. This hydrophobic character of bile also becomes important in the metabolism of highly non-polar drugs.
The liver holds approximately 10-15% of the total blood supply at any moment, and all blood that departs the stomach or intestines passes through the liver enabling absorption of nutrients (or metabolism of xenobiotics in the first-pass effect ). The liver is comprised of functional units termed lobules, which are typically hexagonal structures characterized by a hepatic portal vein, artery, and bile duct at each hexagonal vertex. The portal veins drain into a central vein at the focal point of the hexagonal lobule, that carries blood away from the liver into systemic circulation. These veins (as well as the ducts and arteries) are lined by parenchymal liver cells which are called hepatocytes . Hepatocytes comprise approximately 60-70% of all the cells in the liver and have an asymmetric cellular structure with the periportal side (Zone 1) facing oxygenated blood/nutrients. The cellular face furthest away from the portal triad is called Zone 3 and plays the largest role in detoxification and biotransformation of drugs. The space sandwiched between these zones (pericentral region) is intuitively referred to as Zone 2 of the hepatocyte.
Importantly, the flow of blood through the veins and the flow of bile through the ducts occur in opposite directions to each other. This is consistent with the generation of both substances as bile is produced in the liver, whereas blood enters the liver for perfusion. As mentioned, blood drains from the branches of the hepatic veins into the centre vein of the lobule.
5.2 Phase I Metabolism
Drug metabolism refers to the chemical modification of a drug, and the overall goal is to generate a more polar or water-soluble derivative that is amenable to elimination. Drug metabolism is generally classified as either Phase I or Phase II, although these do not need to be successive steps, and some drugs will only be processed through one of these two phases of metabolism. Both phases of metabolism occur in the hepatocytes of the liver.
Phase I metabolism is characterized by functionalization of the drug via one or more of the reactions:
- Oxidation (electron removal, dehydrogenation, or oxygenation)
- Reduction (electron addition, hydrogenation, or removal of oxygen)
- Hydration/dehydration (hydrolysis, and addition or removal of water)
These transformations are carried out in the endoplasmic reticulum of hepatocytes and the most prominent Phase I reactions are oxidations. In an ex vivo setting, hepatocyte cells can be disrupted and processed by equilibrium density centrifugation to isolate vesicle-like pieces of the endoplasmic reticulum which are called microsomes . Although microsomes do not exist naturally, they provide a convenient tool to analyze Phase I metabolism. Over 75% of Phase I metabolism is performed by a class of enzymes called Cytochrome P450 (CYPs or P450) enzymes. These enzymes are heme-coupled monooxygenases and they bind oxygen (as well as carbon monoxide which leads to a wavelength absorption at 450 nm and serves as their namesake).
5.2.1 Cytochrome P450 Enzymes
There are at least 57 CYP enzymes encoded within in the human genome, with CYP3A4 and CYP2D6 having the highest abundance and contributing to >50% of all CYP-related metabolism. The nomenclature for these enzymes is based on the prefix CYP followed by an Arabic numeral indicating the protein family (usually >40% sequence identity), followed by a letter for the sub-family (>55% sequence identity), and the final number for the isozyme (often greater than 90% sequence identity). Importantly, genetic polymorphisms between CYP family members can result in differential processing of drugs between different genetic populations. For example, the drug diazepam (anxiety and seizure medication) is primarily metabolized by CYP3A4 and CYP2C19. However, mutations in 2% of Europeans, 14% of East Asians, and 57% of Oceanians result in deficient/absent CYP2C19 activity which leads to prolonged effects of the drugs in these patients. Poor drug metabolism can lead to an exaggerated pharmacological response, and the longer lifetime of the compound can also increase toxicity. Conversely, super-fast or rapid metabolizers (individuals with highly active CYP isoforms) can have altered drug responses by due to substantially lowered active drug lifespan. In both cases, clinical implications of polymorphisms require altering the dosages for the therapeutic regimen.
Since multiple drugs converge on CYP enzymes for metabolic processing, these oxygenases are also responsible for a number of drug-drug interactions. Several drugs are either substrates for CYP enzymes or act as inhibitors (prevent a substrate from engaging the CYP enzyme) or inducers (promote activity or gene synthesis of the CYP enzyme). For example, saquinavir (a protease inhibitor used for HIV treatment) is a substrate for CYP3A4, but can be administered with ritonavir (a CYP34A inhibitor) which enables a 33% increase in peak plasma concentrations of saquinavir. Similarly, serotonin uptake inhibitors such as fluoxetine inhibit CYP2D6. CYP2D6 processes codeine to morphine and a patient prescribed these inhibitors would therefore not benefit from the analgesic properties of codeine. There are also multiple inducers, for example CYP1A2 bio-transforms carcinogenic chemicals from cigarette smoke and is also induced by these agents in a positive feedback cycle. CYP profiling to determine which enzymes process a newly developed drug is important to determine any contradictions and metabolism concerns.
The mechanism for CYP oxidation is based on free radical chemistry that leverages different oxidation states of iron to facilitate electron transfer between the metal centre bound within a porphyrin ring called protoporphyrin IX (Figure 5.1). The overall process involves a drug (CYP substrate) with a potentially oxidizable bond (X-H) binding the low-spin Fe 3+ (one unpaired electron S = ½, with a hexacoordinate metal centre including one water molecule) leading to a high-spin Fe 3+ (five unpaired electrons S = 5/2, pentacoordinate with no water, and a shortened Fe-S bond , pulling the iron slightly below the plane of the heme). The drug substrate binding also triggers reduction of Fe 3+ to Fe 2+ with an electron originating from NADPH. The reduced Fe 2+ has a high affinity for diatomic gasses such as O 2 (and also CO) and transfers the electron to generate a superoxide Fe 3+ complex. The Fe 3+ ion picks up an additional electron (again originating from NADPH), which is also transferred to the superoxide anion to generate a peroxide anion (O 2 2- ) species that is split by two protons to form one water molecule and an iron-oxo complex called oxene. This oxene species is highly electrophilic and reactive and inserts into the substrate X-H bond yielding the desired oxidized product. This process only superficially describes the crystal field theory required for understanding these redox reactions, and the most relevant aspects for medicinal chemists are in understanding the types of C–H bonds that are susceptible to CYP oxidation and their final products.
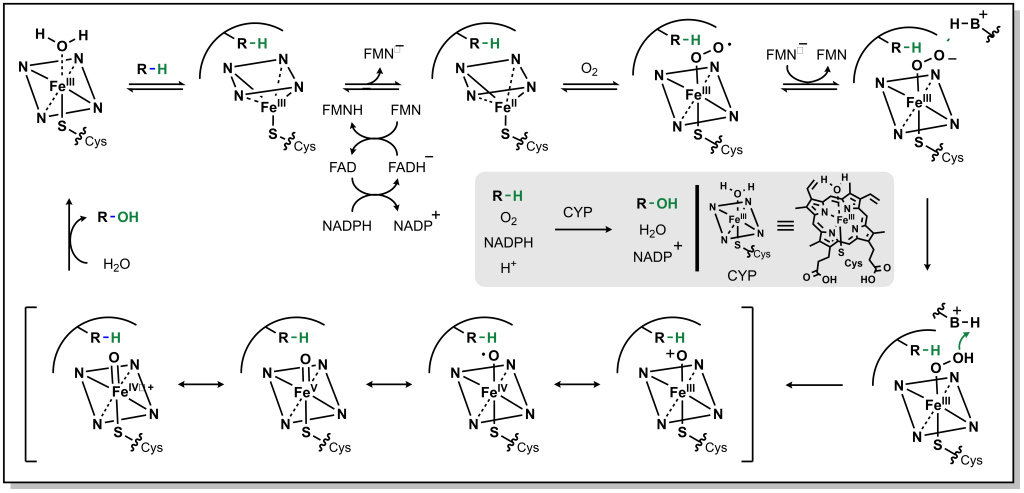
FIGURE 5.1 Reaction mechanism for metabolism via CYP enzymes.
5.2.2 Oxidation of sp 3 Carbon Atoms
5.2.2.1 aliphatic straight chain and ring systems.
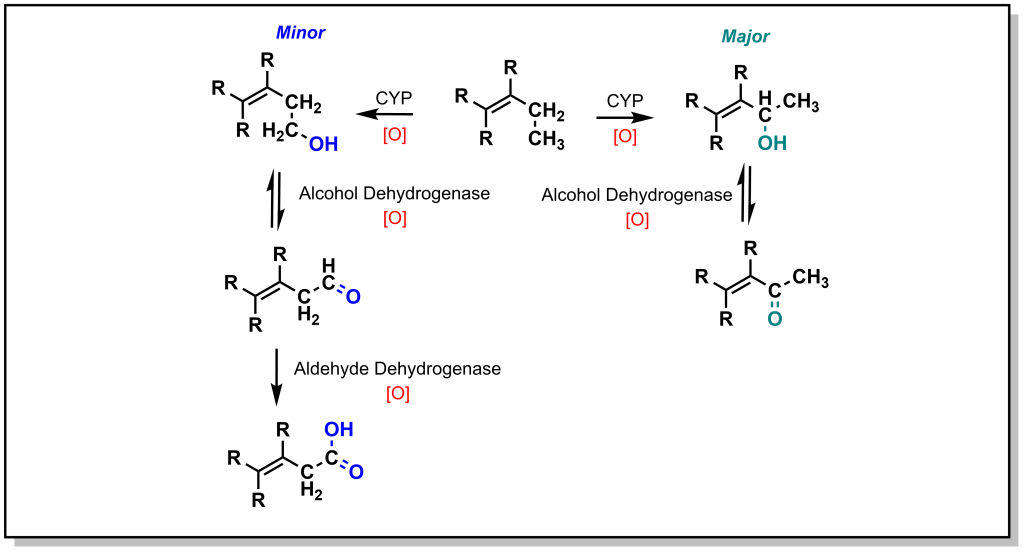
The main mechanism for metabolism of an sp 3 carbon atom is hydroxylation. If there are multiple sp 3 carbon atoms in the drug, there is a preference that hydroxylation will occur at the more substituted carbon atom. This site selectivity arises from the electronics of radical stability. Consider the homolysis of the C-H bond during cleavage in Figure 5.2. The radical generated on the carbon atom is most stable on the ‘internal’ carbon atom and forms the major product. Regardless, the alcohol generated from both the major and minor products will be further oxidized by a non-CYP Phase I enzyme called Alcohol Dehydrogenase (ADH) to give a carbonyl (ketone or aldehyde). For any aldehydes generated as a result of terminal carbon oxidation, the molecule is further oxidized by a comparable Aldehyde Dehydrogenase (ALDH2) to yield a carboxylic acid. Notably, alcohol dehydrogenation is reversible, but the activity of aldehyde dehydrogenase is not reversible.
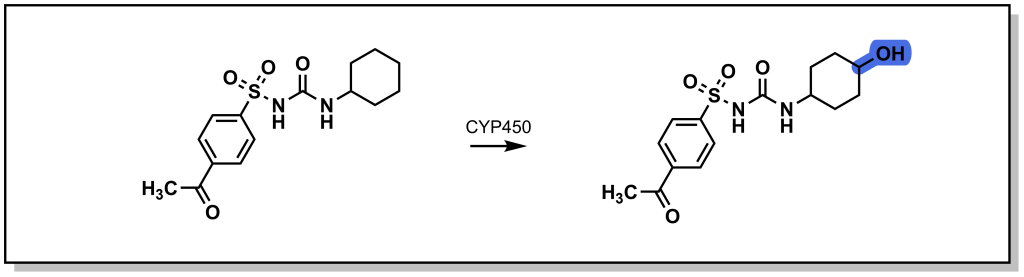
Saturated ring systems are a common functionality in different drug molecules. Due to sterics , oxidation generally occurs at the 3- or 4- ring position as shown with the metabolism of acetohexamide in Figure 5.3. Substitution at the 4- position during SAR design is often considered to impede Phase I enzyme access and sterically block hydroxylation, thereby extending drug half-life.
5.2.2.2 Allylic and Propargylic Systems
As shown with both examples above, both sterics and electronics can play a role in metabolism at sp 3 carbon atoms. As such, there is a site preference of hydroxylation of sp 3 carbon atoms alpha to an unsaturated system. Based on electronic effects, the proximity to a double/triple bond provides stability to a radical (Figure 5.4). Furthermore, in an unsaturated system, the resulting alcohol is dehydrogenated to create a 1,4-conjugated system which has excellent electrophilic properties for Phase II metabolism.
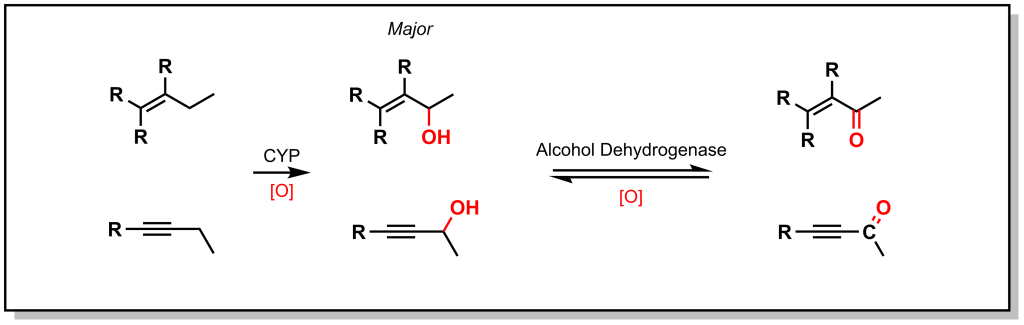
Hexobarbital (a barbiturate derivative used as an anesthetic in the mid-20 th century) contains a cyclohexene substituent (Figure 5.5). Based on sterics, metabolism could be favoured at the 4-position. However, hexobarbital is hydroxylated by CYP2B1 at the 3- position, alpha to the double bond (due to increased electronic stability of the radical). The resulting alcohol is either oxidized to an α,β-unsaturated ketone or is a substrate for UDP-glucuronosyltransferases (discussed in later sections on Phase II metabolism).

5.2.2.3 Halogenated Systems
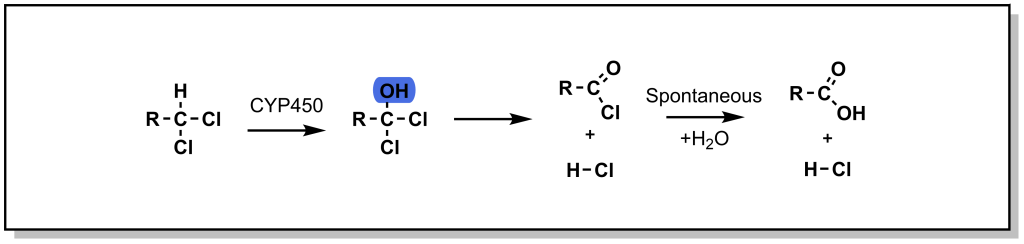
In the case of a halogenated sp 3 centre, oxidation (hydroxylation) can lead to a high-energy / unstable intermediate, that can undergo elimination of the halogen to form a ketone. This oxidative dehalogenation is more prevalent in the presence of two halogen atoms on the sp 3 centre. For example, chloramphenicol is an antibiotic that disrupts bacterial protein synthesis. Chloramphenicol possesses a terminal di-chloro moiety that is oxidized to an acid halide intermediate (very reactive) and further reacts to form a terminal acid moiety. Note that oxidative dehalogenation is not feasible with fluorinated carbon atoms, as the C-F bond is extremely strong and cannot be homolyzed by CYP enzymes. In this way, fluorine can function as a bioisostere for hydrogen.
5.2.3 Oxidation of sp 2 and sp Carbon Atoms
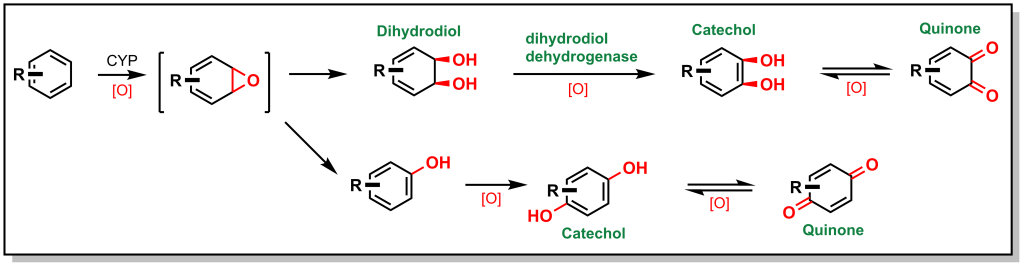
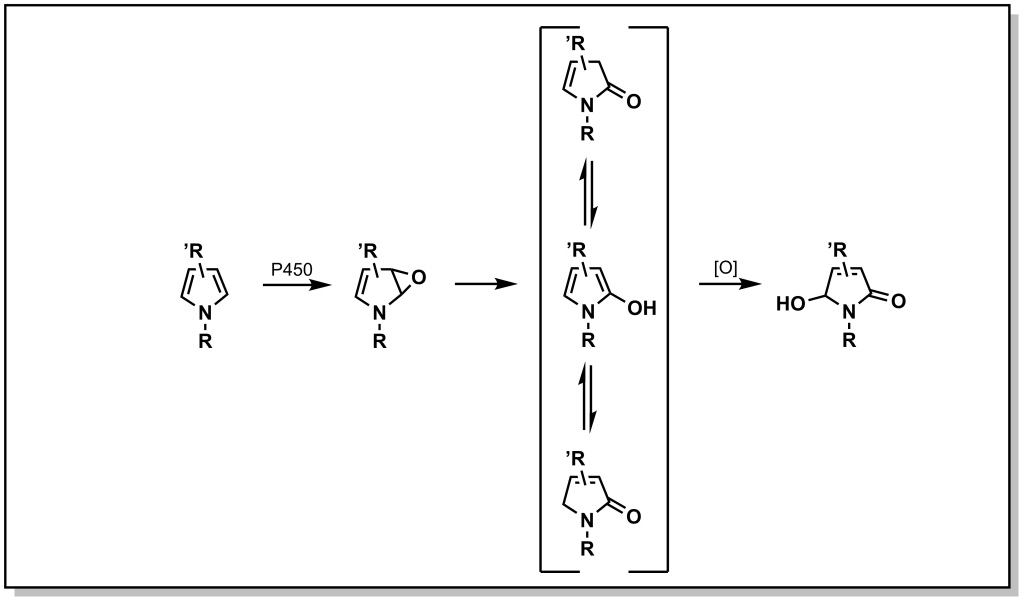
Unsaturated carbon scaffolds are also susceptible to oxidation by CYPs. For example, the oxygen atom from the CYP enzyme can insert into the π system of benzene to form a highly reactive epoxide (Figure. 5.7). The reactive epoxide is subsequently hydrolyzed by epoxide hydrolase to yield a dihydrodiol or it can re-arrange and re-aromatize to phenol via proton catalysis. In either case, these substrates are further oxidized to catechol (bis-phenol) derivatives. These catechols are substantially more polar and can be directly excreted, or oxidized by dehydrogenases to yield reactive quinones (which are substrates for Phase II metabolism).
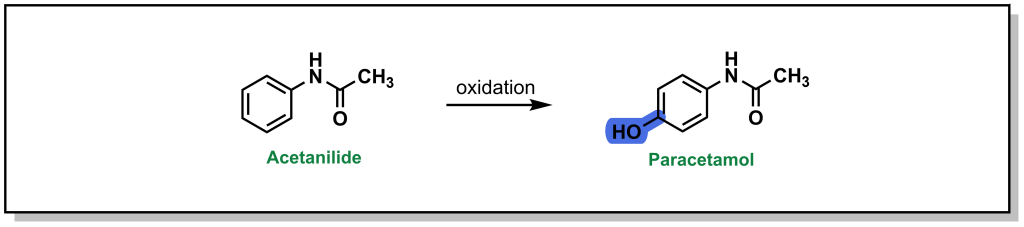
Acetanilide is a discontinued drug that was the first aniline derivative identified to possess analgesic properties. These favourable therapeutic properties are a result of aromatic hydroxylation at the para-position ( sp 2 carbon) which gives paracetamol (commonly known as acetaminophen or Tylenol). (Figure 5.8) However, there is also a minor metabolic product of N -dealkylation (discussed below) which results in aniline and is nephrotoxic leading to hemolytic anemias and other safety concerns.
Compared to sp 3 oxidation, the metabolic modification of sp 2 and sp carbons is a significantly slower process. Furthermore, it is also uncommon in modern drug discovery to observe free, unsubstituted phenyl rings in drug structures. This is partly because common strategies for bypassing sp 2 metabolism involve generating more electron deficient rings or adding a fluorine or chlorine/substituent to the phenyl ring.
The utility of benzene in synthetic organic chemistry represents an interesting example of differences in drug metabolism of sp 3 and sp 2 carbons. As a simple aromatic liquid, benzene was routinely employed as an organic solvent for a number of industrial applications as well as organic synthesis. However, it is highly toxic and has been largely superseded by toluene, which still has safety concerns, but is far less toxic than benzene. This difference arises from the sp 3 benzylic position of toluene which is much weaker (C-H bond strength = 85 kcal/mol) than the aryl C-H bond (110 kcal/mol). As such the benzylic carbon is more readily oxidized by CYP enzymes, creating a benzylic radical that is further dehydrogenated and oxidized to afford benzoic acid, which is rapidly cleared. Conversely, oxidation of the benzene sp 2 carbon is substantially slower, and therefore benzene remains in the body for longer time periods. Moreover, the toluene oxidation metabolite has limited safety challenges, whereas benzene oxidation leads to a reactive quinone electrophile which is an excellent 1,4-Michael acceptor.
5.2.4 Oxidation of Hetero-atoms Bound to Carbon Atoms
Hetero-atoms (O, N, and S) are common in drug discovery and are often responsible for key target-drug interactions. The general strategy employed by Phase I metabolism enzymes is to either i) oxidize the hetero-atom directly (referred to as heteroatom oxygenation ) or ii) oxidize the alpha-carbon next to the hetero-atom (often referred to as de-alkylation although there is an initial hydroxylation step; Figure 5.9).
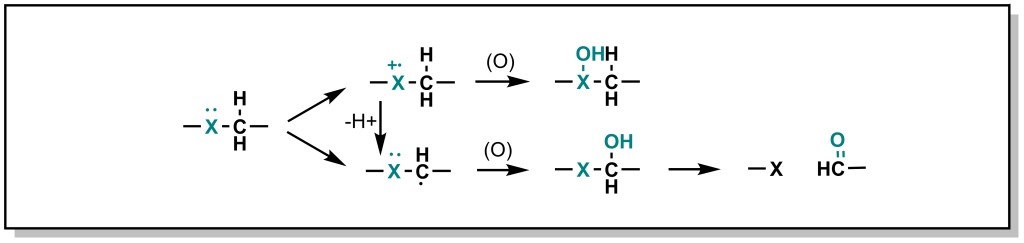
In heteroatom oxygenation, there is an initial single electron transfer (SET) from the heteroatom to the iron-oxygen complex of the CYP enzyme. This loss of an electron creates a radical cation intermediate on the heteroatom that eventually reacts with an oxygen provided by the CYP enzyme. In de-alkylation, there is an initial hydrogen atom abstraction (HAT) from the alpha-carbon which ends up on the iron-oxygen complex of the CYP enzyme (becoming iron-hydroxyl). This loss of a hydrogen atom creates a neutral carbon radical intermediate that eventually reacts with the hydroxyl provided by the CYP enzyme. The type of metabolism (heteroatom oxygenation or de-alkylation via SET or HAT respectively) that occurs depends on i) the hetero-atom and ii) the substituents of the heteroatom.
- For carbon-oxygen systems, heteroatom oxygenation does not occur because removing a single electron to generate a positively charged oxygen is highly energetically unfavorable. Therefore, for carbon-oxygen systems, metabolism occurs via HAT and de-alkylation.
- For carbon-nitrogen systems, HAT and dealkylation are highly favoured (similar to carbon-oxygen systems above). However, SET is not as unfavourable for nitrogen species and therefore heteroatom oxygenation is feasible and can proceed (usually as a minor product). SET/Hetero-atom oxidation is more favorable when the radical cation is destabilized based on the nearby substituents, or if there are no alpha-protons available for abstraction.
- For carbon-sulfur systems, SET and heteroatom oxygenation is energetically favored due to the formation of a stable sulfur radical cation.
5.2.4.1 Metabolism of O -Containing Moieties
5.2.4.1.1 metabolism via o -dealkylation.
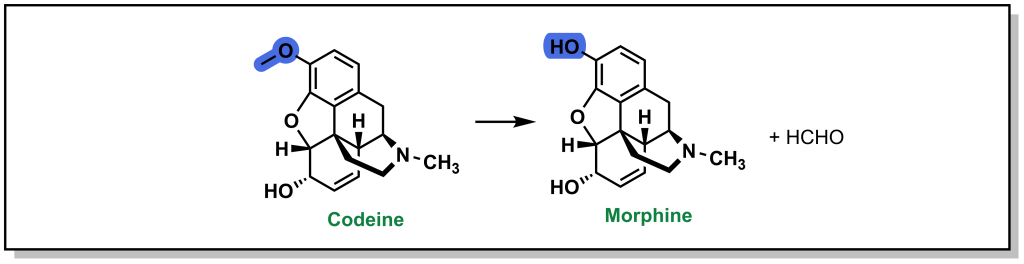
As mentioned above, the carbon-oxygen containing moieties will only undergo de-alkylation and will not be directly oxidized on the oxygen atom. These C-O moieties are commonly found as ethers in drugs and will metabolize to an alcohol with release of a ketone or aldehyde. As with all metabolic reactions, steric hindrance of the ether can block metabolism. A well-known example of O -dealkylation involves metabolism of codeine to morphine where the terminal ether is metabolized and also releases the parent phenol (Figure 5.10). Notably, this only accounts for ~10% of the modification of ingested codeine. Approximately 10% of codeine is also N -dealkylated (which will be discussed in the next section), and the remaining 80% is directly glucuronidated on the cyclohexenyl alcohol, in Phase II metabolism. Notably, the alternative C-O linkage in the ring system is not metabolized due to steric hindrance.
5.2.4.2 Metabolism of N -Containing Moieties
5.2.4.2.1 metabolism via n -dealkylation.
The most common types of nitrogen containing compounds in drug discovery are tertiary amines (including both aliphatic and aromatic amines). Oxidation of these amines predominantly occurs as discussed above, where a carbon atom alpha to the nitrogen is hydroxylated to form a carbinolamine and this intermediate rearranges via cleavage of the C-N bond ( de-alkylation ; Figure 5.11).
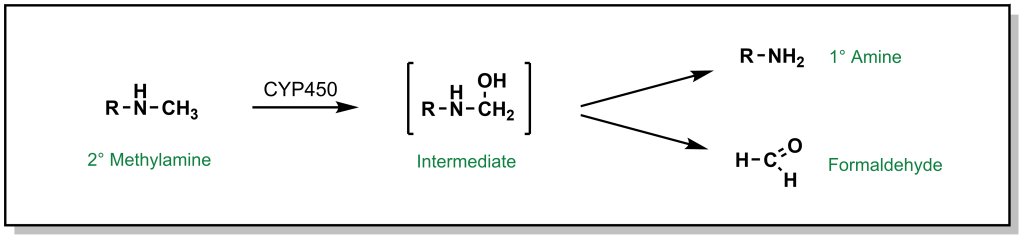
FIGURE 5.11 Oxidation of secondary amines can lead to N-dealkylation.
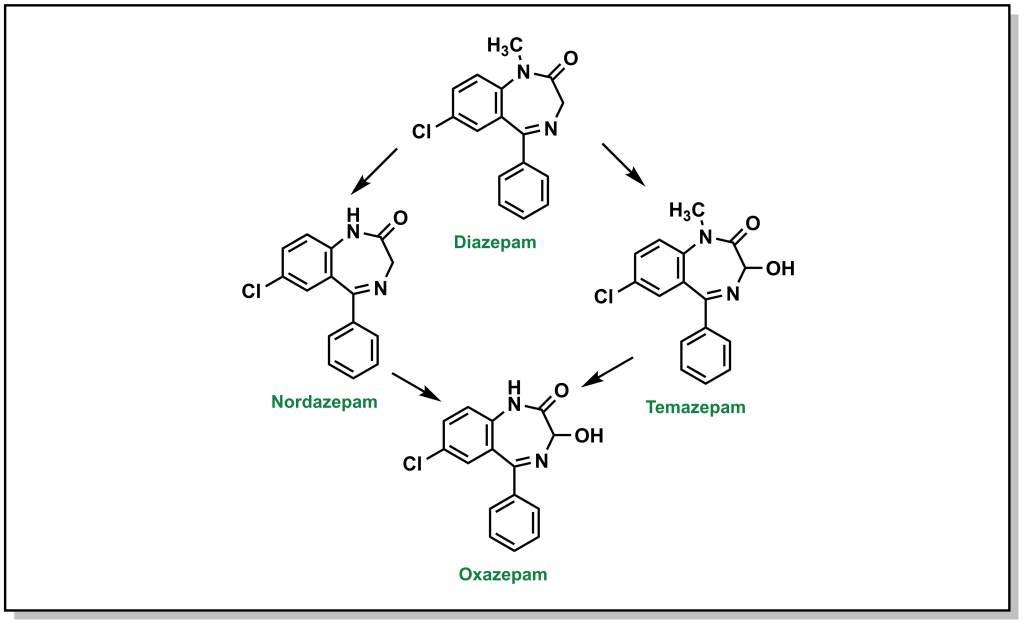
For example, the tertiary amine of diazepam is N -dealkylated in Phase I metabolism (Figure 5.12). Additional metabolic steps of diazepam include hydroxylation of the sp 3 carbon which becomes an available substrate for glucuronidation.
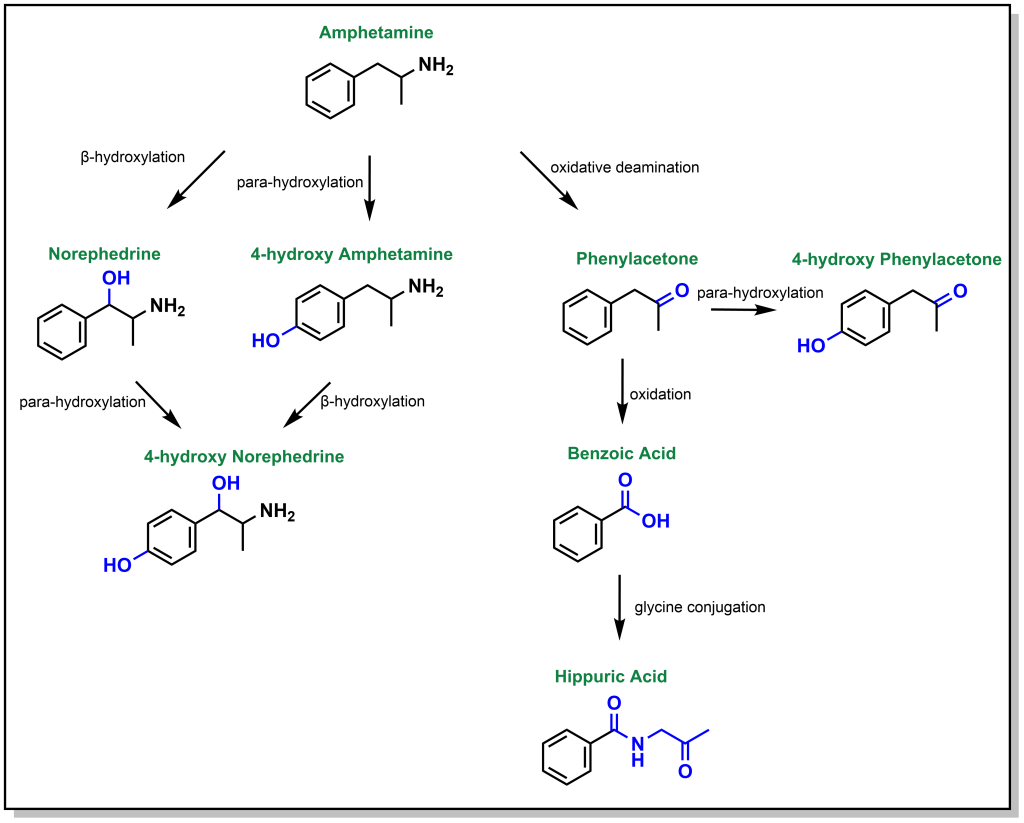
Dealkylation occurs for tertiary and secondary amines. Primary aliphatic amines are metabolized via the same mechanism, where hydroxylation of the alpha-carbon occurs, followed by rearrangement to release ammonia, and this is termed de-amination (as opposed to N -dealkylation). Amphetamine is a respiratory and CNS stimulant that is metabolically processed by different reactions including an oxidative deamination but also β-hydroxylation (of the sp 3 benzylic carbon) and para-hydroxylation (of the sp 2 phenyl ring; Figure 5.13).
5.2.4.2.2 Metabolism via N -Oxidation
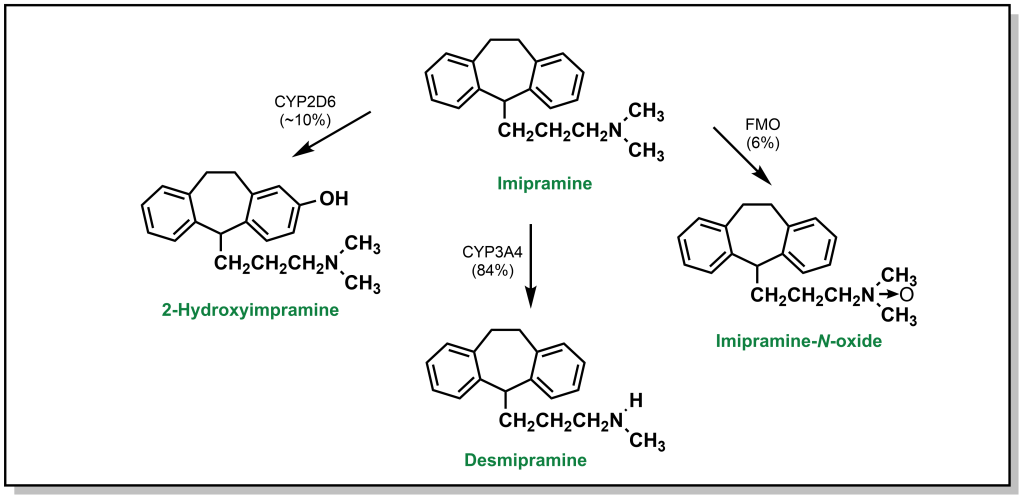
In addition to de-alkylation and de-amination, tertiary amines can undergo metabolic N -oxidation by a group of Phase I enzymes called flavin-containing monooxygenases (FMO) which enable oxidation on the heteroatom to yield N -oxide products. As mentioned, this is generally less favourable due to initial formation of a radical cation on the nitrogen atom. For instance, imipramine is an amine-based tricyclic antidepressant. Its tertiary amine is predominantly processed by hepatocytes through N -dealkylation via CYP3A4 (84%). Para-hydroxylation by CYP2D6 accounts for ~10% of the metabolic products observed. Finally, the remaining 6% is metabolized to an imipramine- N -oxide via FMO (Figure 5.14).
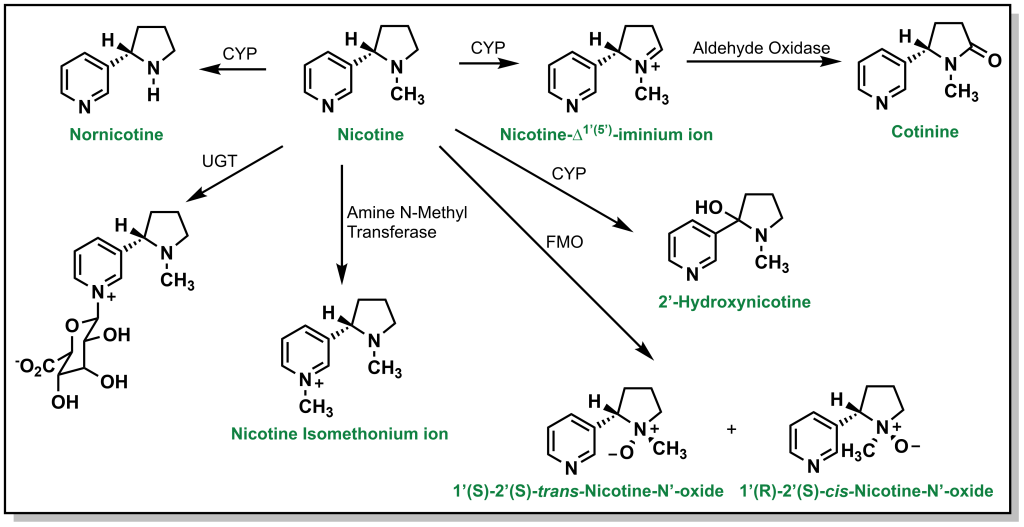
N -Oxidation can also occur with alicyclic amines such as nicotine, which harbours a nitrogen in the pyrrolidine and pyridine rings. Approximately 10% of nicotine is N -dealkylated at the pyrrolidine and ~75% is oxidized to cotinine (amide) via a similar hydroxylation mechanism. The pyridine can also be directly glucuronidated through Phase II processes (5%). Additional metabolism occurs through FMOs to create nicotine n-oxide (Figure 5.15).
Nitrogen–containing aliphatic, and heteroaromatic ring systems are extremely common substructures in many drugs. Indoles and pyrroles represent electron-rich heterocycles which helps to facilitate metabolism via ring oxidation, with a preference towards oxidation on carbon atoms adjacent to the nitrogen. Metabolism trends from the analysis of several drug libraries indicate that a greater number of nitrogen atoms in the ring, reduce the possibility of Phase I oxidations. Tetrazoles in particular, are relatively inert to oxidation (and are important bio-isosteres of carboxylic acids, discussed later). Importantly, a number of nitrogen containing heterocycles including pyridines, imidazoles, and triazoles can engage with the heme-group to block activity of CYP enzymes. However, they can also be excellent substrates for Phase II metabolism, especially N -glucuronidation as observed with nicotine (Figure 5.16).
5.2.4.3 Metabolism of S -Containing Moieties
5.2.4.3.1 metabolism via s -oxidation.
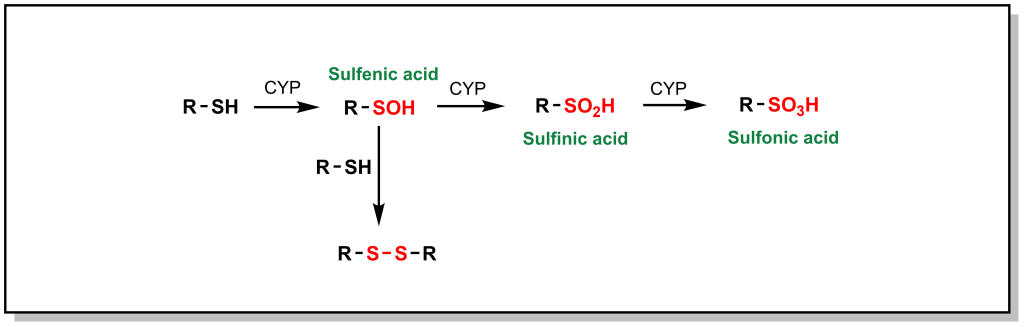
Phase I metabolism for sulfur containing compounds generally proceeds via direct oxidation of the heteroatom (SET), which impacts different sulfur containing functional groups including sulfides (especially in ring structures), free thiols, sulfonyls, and thioketones. Free thiols are uncommon in modern drug molecules due to their reducing potential, which enables them to form di-sulfide bridges with cysteine residues or self-aggregate. However, in such drugs, the thiol is directly hydroxylated to form sulfenic acid, which is further oxidized to sulfonic acid (Figure 5.17).
Higher oxidation states of sulfur, such as sulfonyls found in sulfonylureas and sulfonylamides, are significantly more hydrophilic, which facilitates their elimination. Thioethers (sulfides) can be oxidized to the corresponding sulfoxide and then further oxidized to the sulfone. For example, thioridazine (an antipsychotic medication used for schizophrenia) contains two sulfides and both of these sulfides can be S -oxidized (additional metabolic pathways include N -dealkylation; see Figure 5.18a). Thioketones undergo desulfuration via a similar oxidation of the sulfur atom as seen with thiopental, which produces a carbonyl-containing product (Figure 5.18b).
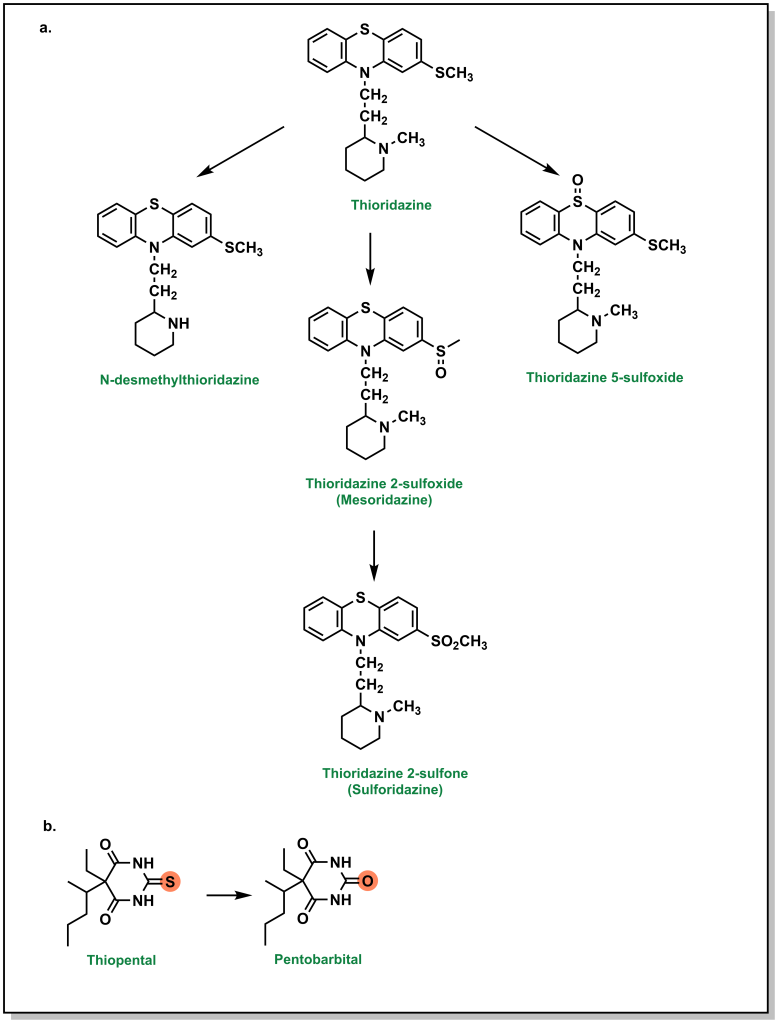
Importantly, metabolism of sulfur atoms has been associated with bioactivation and generation of reactive metabolites that are associated with toxicities, and understanding metabolite profiles can be important when selecting sulfur functional groups.
5.2.5 Other Phase I Oxidation Enzymes
In addition to CYPs, there are other non-CYP enzymes involved in Phase I metabolism, some of which have been highlighted above. The most commonly known non-CYP Phase I enzyme is Alcohol Dehydrogenase , which oxidizes alcohol functional groups into aldehydes or ketones, and Aldehyde Dehydrogenase , which oxidizes aldehydes to carboxylic acids. These enzymes are found exclusively in hepatocytes and have well characterized functions on ethanol, methanol, and ethylene glycol. Other important enzymes are Flavin Containing Monooxygenases (FMOs), which operate in a similar manner to CYPs and yield highly similar by-products. However, while CYPs utilize the high-reactive oxene as an oxidizing agent, FMOs use slightly less powerful peroxides. As such, FMOs can only oxidize heteroatoms and cannot oxidize primary amines, highly charged or polyvalent species.
Regardless of the oxidation mechanism, the overall goal of Phase I enzymes is to increase solubility (hydrophilicity) or expose a functional group for conjugation in Phase II.
5.2.6 Reductions
5.2.6.1 carbonyls.
Reductions are less common since they usually mask reactive groups, but they are largely involved in reducing carbonyls (mainly ketones) to hydroxy groups, which are more susceptible to conjugation reactions (Phase II). The order for reactivity generally follows the principles of sterics and electronics, namely aliphatic ketones/aldehyde > aromatic (benzylic) ketones/aldehyde > esters, acids, and amides. Importantly, the reduction of ketones can lead to the generation of stereocentres and stereoisomeric alcohols. For example, one of the routes for the metabolism of the blood thinner warfarin involves the reduction of its ketone by Human Hepatic Cytosolic Reductase to yield two diastereomeric alcohols.
5.2.6.2 Nitro- and Azo- Compounds
Nitro- reductions are performed by NADPH-dependent microsomal and soluble nitroreductases, whereas NADPH-dependent multicomponent hepatic microsomal reductase systems reduce azo- groups. The end products of these reactions are primary amines (Figure 5.19). Bacterial reductases in the gastrointestinal tract have the capacity to reduce both nitro and azo groups.
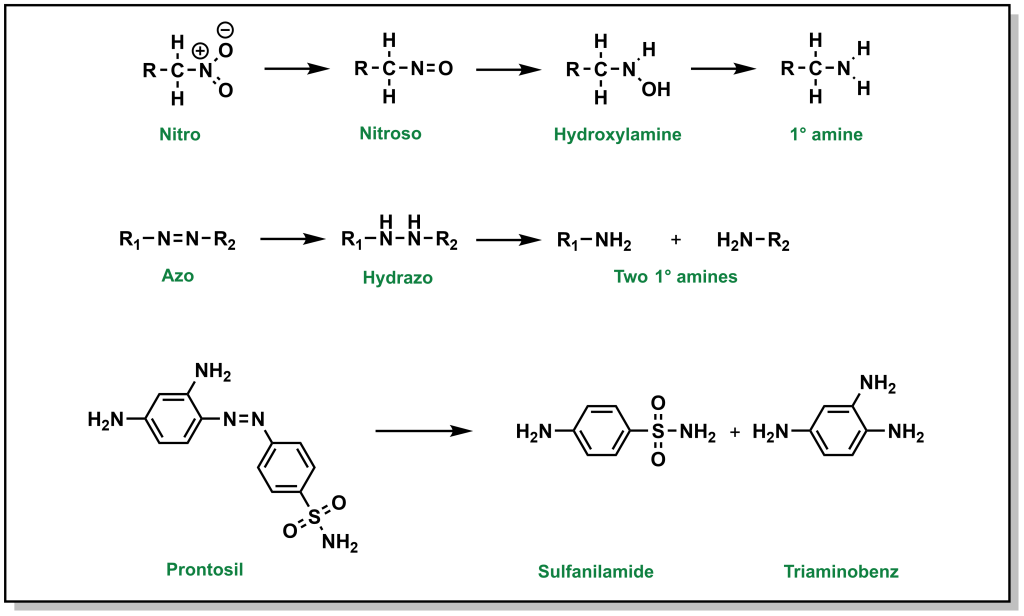
5.2.7 Hydrolysis
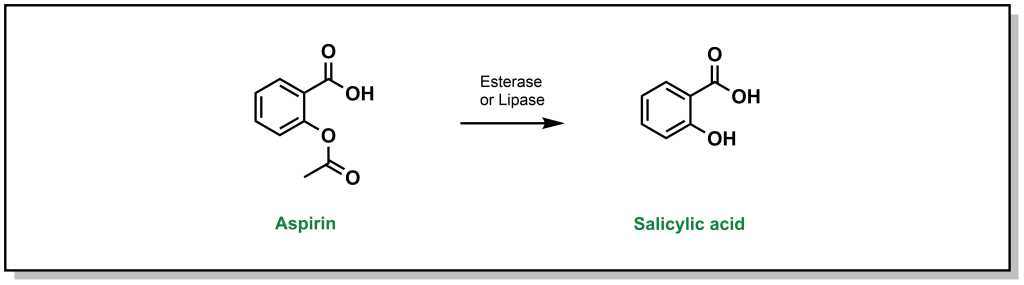
Hydrolysis reactions are fundamentally distinct from previous Phase I reactions described above, in that there is no change to the redox level of the drug molecule, and there is usually a substantial change to original substrate (cleavage of a functional group and generation of multiple products). The reactivity of a substrate towards hydrolysis is determined by the electrophilicity of the carbonyl carbon (or other reactive centre) and the nucleofugality of the leaving group substituents. The rate is found to follow the trend of thioester > ester > carbonate > amide > carbamate. One of the classic examples is hydrolysis of aspirin which can be converted to salicylic acid by esterases. (Figure 5.20)
5.3 Phase II Metabolism
Whereas Phase I is largely focussed on oxidases that aim to introduce a polar functional group, Phase II involves transferases that conjugate a biomolecule onto the drug. Phase I reactions may not necessarily generate a pharmacologically inactive species, but Phase II reactions are regarded as detoxifying and will inactivate drugs to prevent damage to macromolecules or to prepare drugs for efficient elimination. Substrates are covalently coupled to an endogenous molecule, which is generally very polar and large (100-300 Da) and can usurp the physical properties of the drug (LogP, tPSA, etc.). Since these processes can be energetically unfavourable, Phase II transferase enzymes will activate the endogenous molecule to prepare it for conjugation to the drug.
5.3.1 UDP-Glucuronosyltransferases (UGT)
Glucuronidation by UGTs represents the most common Phase II metabolic process and involves the transfer of a glucuronosyl group to either an O -, N -, S – or C – atom. Physiologically, the ubiquity of glucuronidation arises from the availability of D-glucuronic acid (a D-glucose derivative), the enzymatic activation of the conjugate, and the stark changes in water solubility by addition of the glucose-group. In this conjugation reaction, glucose-1-phosphate is first activated to uridine-5-diphosphoate-alpha-glucose (UDPG) by the action of a phosphorylase. The 6-C is oxidized to an acid via UDPG-dehydrogenase and two equivalents of NAD + (Figure 5.21a). The glycosidic bond is a high energy bond and easily broken to conjugate onto the substrate via nucleophilic attack of the drug substrate. This results in uridine diphosphate (UDP) functioning as a leaving group. Notably, this SN2 attack of the drug substrate also leads to inversion of stereochemical configuration in the resulting β-linkage to the glucuronic acid. A number of functional groups can participate in this reaction, including weak nucleophiles, and this accounts for approximately one-third of all Phase II metabolism. It is very common to form O -glucuronides via a hydroxy groups (phenolic/alcoholic) or carboxylates (aromatic or aliphatic). For example, morphine is extensively metabolized via O -glucuronidation (Figure 5.21b).
Even carbon atoms, in a minority of cases, can be glucuronidated such as in the case of sulfinpyrazone (a drug for the chronic treatment of gout). This occurs because the H-atom of the tertiary carbon is acidic and susceptible to removal by a base to create a nucleophilic carbanion that undergoes reaction with UDP-glucuronic acid (UDPGA) (Figure 5.21c). As described in previous examples, N-atoms and S-atoms can also be glucuronidated.
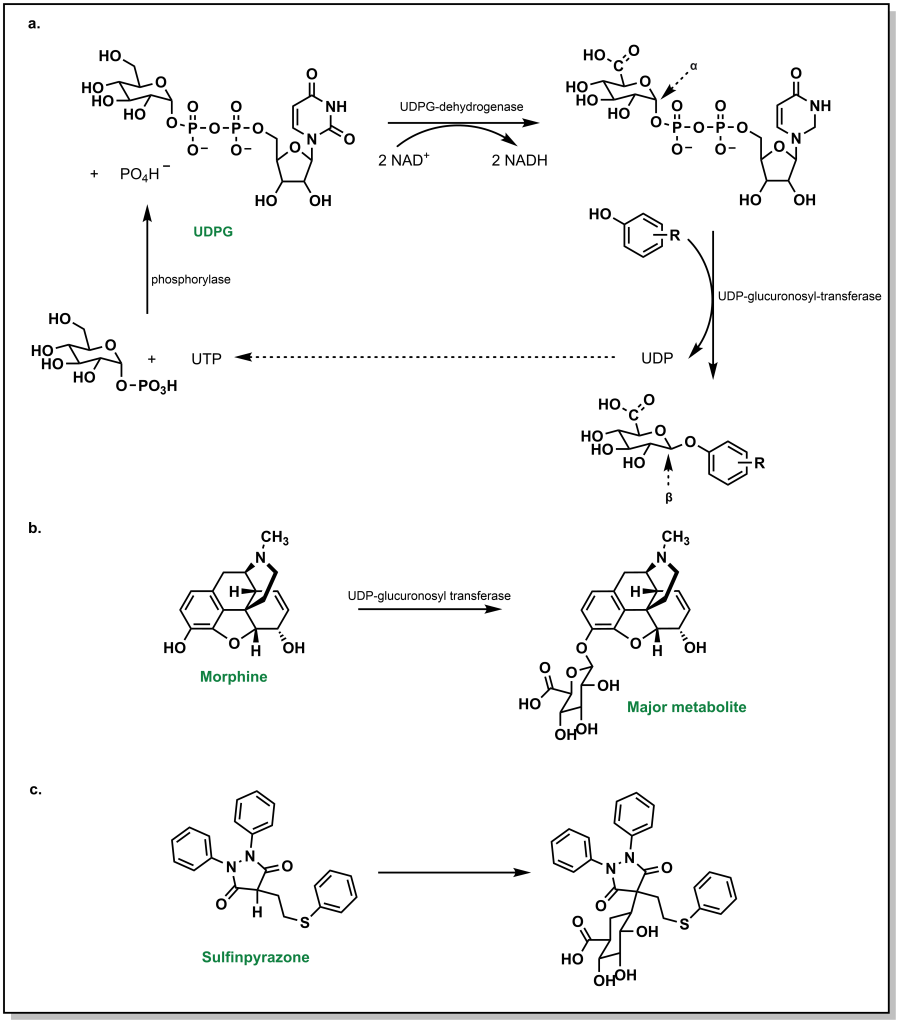
5.3.2 Glutathione-S-Transferase
Glutathionylation is the second-most common Phase II metabolism pathway, and also forms a significant aspect of first-pass metabolism. Glutathionylation is thought of as a highly protective process with de-toxifying effects. Glutathione is a tri-peptide analogue of glycine, cysteine, and glutamate, with a γ-peptide linkage between the carboxyl side chain of glutamate and the alpha amino group of cysteine. The reactivity of glutathione emerges from the cysteine side chain, which has an expected pKa of 8.7 (more acidic than a similar alcohol functionality such as serine, due to stabilization of the negative charge on a larger, polarizable sulfur atom). However, the nucleophilic character can be further enhanced by the action of the enzyme Glutathione S-transferase , which can increase the acidity by 2-3 pKa units, generating a charged thiolate anion. Any drug that has mildly electrophilic sites will be vulnerable to nucleophilic attack. The most common substrates of glutathione-S-transferases are soft electrophiles/Michael acceptors.
- α,β-unsaturated carbonyls (most common electrophile)
- quinones/quinone imine (often formed on Phase I metabolism)
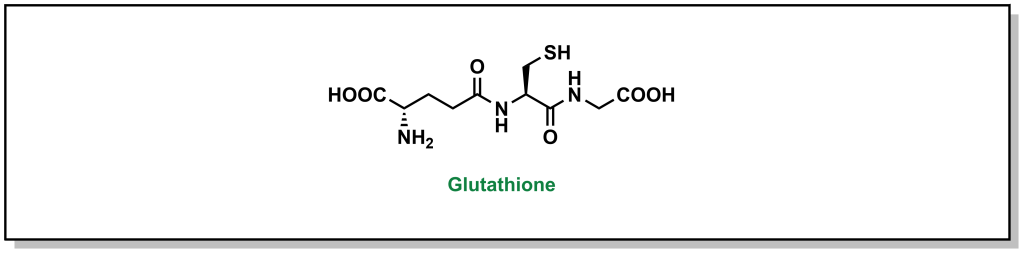
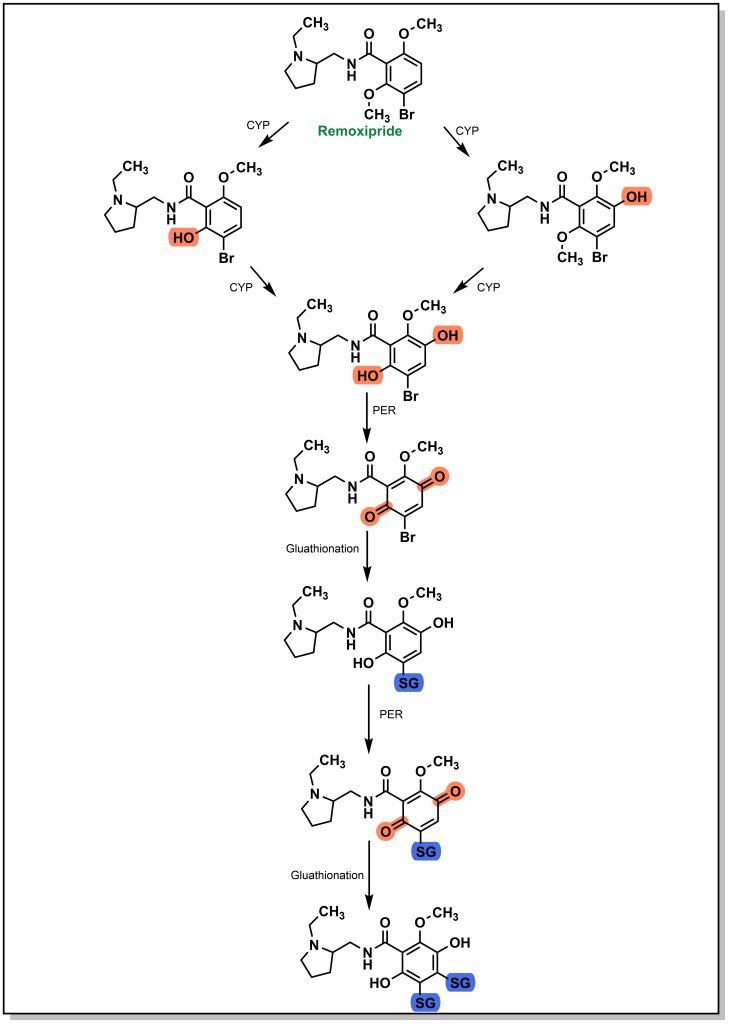
There have been a number of drug examples discussed above that are subject to glutathionylation, such as paracetamol (Tylenol). One case involves remoxipride, which is a substituted benzamide and was previously employed as an antipsychotic for treatment of schizophrenia. As expected, initial CYP metabolism leads to dealkylation of an ether linkage that is further oxidized to a quinone species (Figure 5.23). The presence of a bromo-group enables two glutathione transfer reactions, which inactivates the molecule and prepares it for elimination. Remoxipride was discontinued after 8 cases (2 fatal) of aplastic anemia were detected.
5.3.3 Sulfonyltransferases
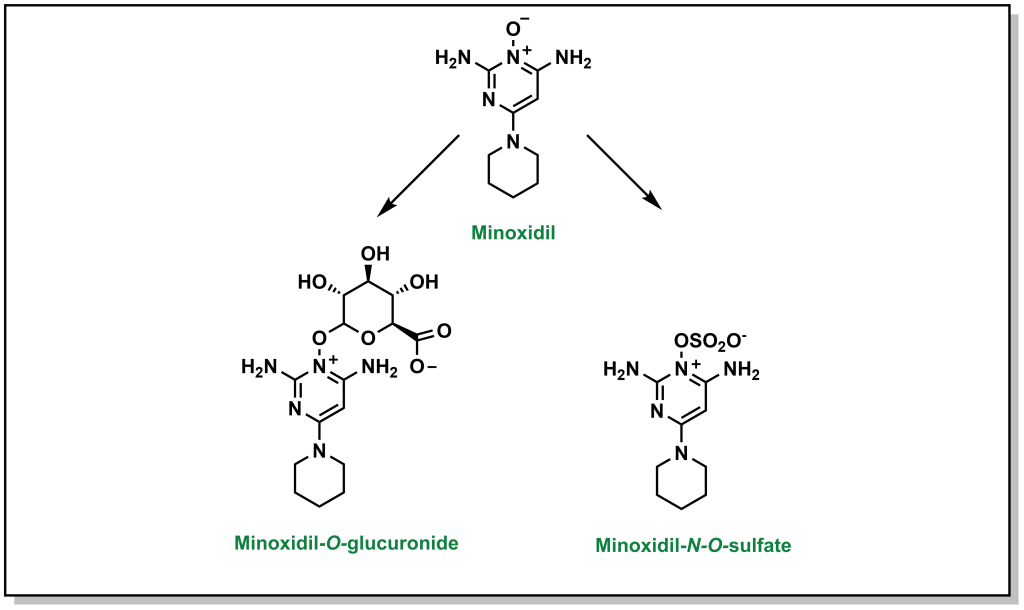
Sulfonylation involves the transfer of sulfate from an endogenous biomolecule to a drug (containing a nucleophilic hetero-atom). Similar to the above Phase II reactions, the introduction of a sulfate markedly alters the hydrophilicity. The sulfate group is provided by a molecule called 3′-phosphoadenosine 5′-phosphosulfate (PAPS) which contains a mixed sulfate/phosphate anhydride. This is a high energy anhydride that is significantly more reactive than di- or tri-phosphates, but has significantly reduced cellular availability compared to glutathione or UDP-glucose. This reaction predominantly occurs with phenols, but is known to occur with alcohols, aryl amines, and N -oxide compounds. For example, minoxidil (Rogaine) is drug used to help with hair loss in men and can be metabolized by the action of sulfonyltransferases (as well as by glucuronidation; Figure 5.24).
5.3.4 Methyl Transferases
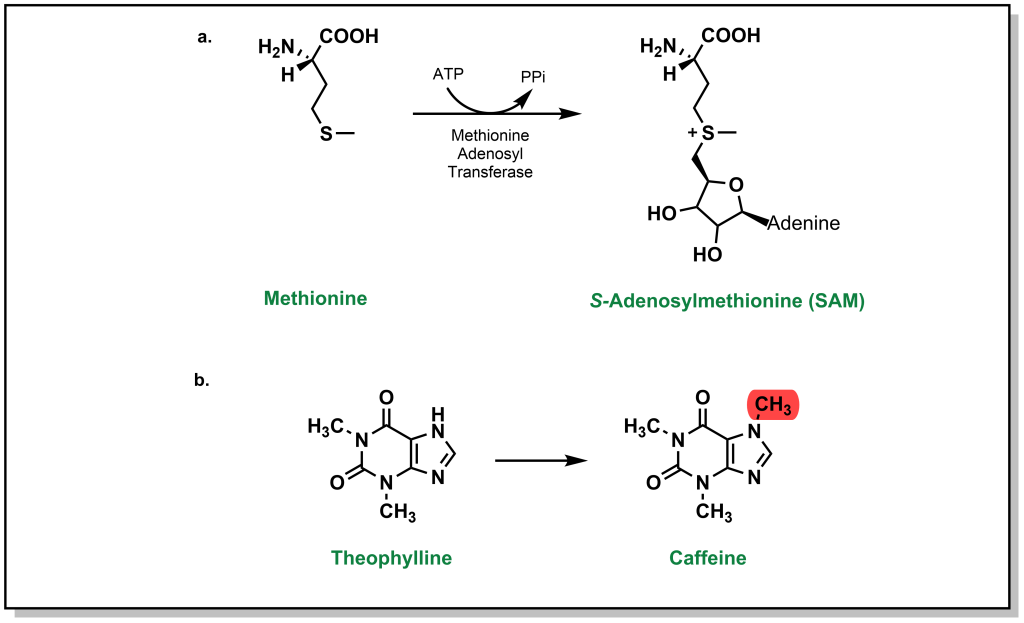
While glucuronidation, glutathionation, sulfonylation, as well as several other Phase II metabolism pathways increase the water-solubility of drugs, processes such as methylation (and acetylation below) do not substantially alter values such as LogP. Instead, these processes are tailored more toward inactivating a drug (an exception to his occurs with the formation of a charged, quaternary ammonium derivative). Any heteroatom with a hydrogen atom is prone to methylation, and this usually occurs at a late stage in the metabolism process. Methylation is carried out by S -adenosylmethionine (SAM) which involves an ATP-activated methyl group from methionine). For example, theophylline is used to treat apnea and breathing issues (which can be common in infants, especially premature babies), but the methylated metabolite is caffeine, which can lead to adverse effects (Figure 5.25).
5.3.5 Acetyl Transferases
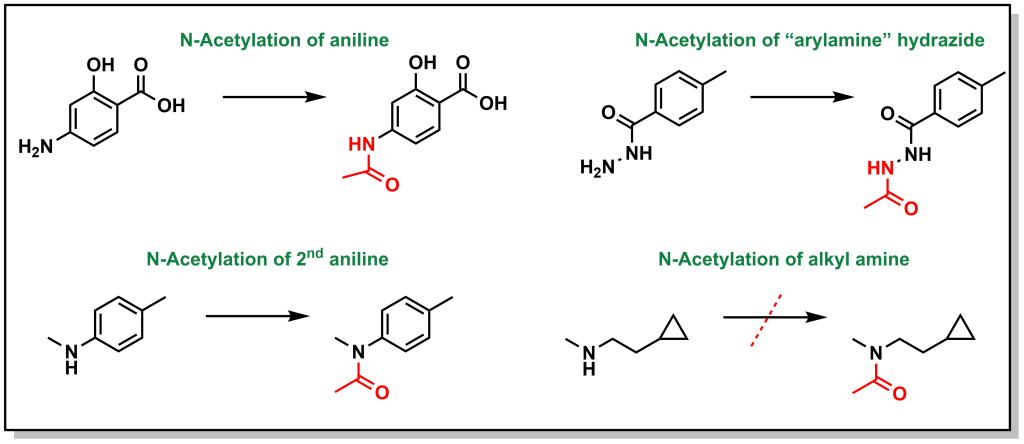
Similar to methylation, acetylation does not substantially alter lipophilicity but facilitates drug inactivation particularly on nucleophilic atoms (N and O). The main substrates are arylamines and sulfonamides, and to a lesser extent hydrazines, hydradizes, and aliphatic amines. The major enzyme system that acetylates the drugs is arylamine N -acetyltransferase . Primary and secondary aliphatic amines are only marginally acetylated (Figure 5.26). This reaction is particularly important in sulfonamide metabolism because acetyl-sulfonamides have reduced solubility and can lead to precipitation, especially in the kidneys, leading to renal toxicities
5.4 Drug Metabolism Exploits Ubiquitous Themes in Chemistry
One of the remarkable features of hepatocytes and drug metabolism within the body is that there is a high efficiency and capacity to metabolize foreign molecules with a wide range of chemical diversity. Instead of developing a tailored nucleophile to react with specific drug, general mechanisms are used to oxidize generic scaffolds found in all drug molecules. This greatly simplifies the extent of biological tools needed and improves genetic efficiency for drug metabolism while maintaining a broad reactivity and substrate scope.
Furthermore, not every single oxidation, reduction, hydration, etc., that can occur will occur. The above examples outline potential reactions with sp 3 and sp 2 carbons, heteroatoms, acids, and bases. Many drugs contain multiple functional groups, and there is usually a preference for specific metabolite profiles that depends on multiple features including the steric hinderance, electrophilicity, and polarity of the molecule. Once the molecule undergoes an initial modification, it may be sufficiently water-soluble to be expelled without further modification. However, in the same way, a single modification may not be sufficient and there can be multiple modifications that occur.
A key part of medicinal chemistry programs is identifying the metabolic liabilities on the drug – positions that are metabolized. This can help extend the lifetime of the drug or identify Phase I and Phase II metabolites. A metabolite identification ( MetID ) experiment is performed where a compound is subjected to hepatocytes and the resulting products are identified through mass spectrometry. The resulting pathway may not be linear and may demonstrate different transformations occurring at the different stages although they all converge on the common goal of preparing the compound for elimination.
An Introduction to Medicinal Chemistry & Molecular Recognition Copyright © 2024 by Elvin D. de Araujo; Timothy Wright; Bilal Saqib; Jeffrey W. Keillor; and Patrick T. Gunning is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License , except where otherwise noted.
Share This Book
An official website of the United States government
Official websites use .gov A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
- Publications
- Account settings
- Advanced Search
- Journal List
Drug Metabolism: A Half-Century Plus of Progress, Continued Needs, and New Opportunities
F peter guengerich.
- Author information
- Article notes
- Copyright and License information
Address correspondence to: F. Peter Guengerich, Department of Biochemistry, Vanderbilt University School of Medicine, 2200 Pierce Avenue, 638 Robinson Research Building, Nashville, Tennessee 37232-0146. E-mail: [email protected]
Corresponding author.
Received 2021 Oct 27; Accepted 2022 Jun 28; Issue date 2023 Jan.
The systematic study of drug metabolism began in the 19th Century, but most of what we know now has been learned in the last 50 years. Drug metabolism continues to play a critical role in pharmaceutical development and clinical practice, as well as contributing to toxicology, chemical carcinogenesis, endocrinology, and drug abuse. The importance of the field will continue, but its nature will continue to develop with changes in analytical chemistry, structural biology, and artificial intelligence. Challenges and opportunities include toxicology, defining roles of genetic variations, and application to clinical issues. Although the focus of this Minireview is cytochrome P450, the same principles apply to other enzymes and transporters involved in drug metabolism.
SIGNIFICANCE STATEMENT
Progress in the field of drug metabolism over the past 50 years has helped make the pharmaceutical enterprise what it is today. Drug metabolism will continue to be important. Challenges and opportunities for the future are discussed.
Introduction
I appreciate the editors’ invitation to write a Minireview on the 50th anniversary of Drug Metabolism and Disposition . It is a fine journal that fills an important niche in this field. I continue to be a strong advocate of society-based journals managed by working scientists, published for the benefit of scientists, and have served as an associate editor for journals published by the American Society for Pharmacology and Experimental Therapeutics, the American Chemical Society, the American Association for Cancer Research, and the American Society for Biochemistry and Molecular Biology. I also add that this is (about) the 50th anniversary of the Gordon Research Conference on Drug Metabolism, another important entity in our discipline (with meeting delays caused by pandemic issues). A lot has happened in drug metabolism in the last 50 years, and I can only relate so much. The year 2022 also marks two other anniversaries for me. It is hard to believe that it has been 30 years since I received the American Society for Pharmacology and Experimental Therapeutics B. B. Brodie Award in Drug Metabolism. It has also been 20 years since I was awarded a Docteur Honoris Causa (honorary doctorate) from the University of Paris thanks to Professor Philippe Beaune and other friends in France.
As an undergraduate and graduate student, I had not started out to work on pharmaceutical problems or drugs. My interest began to develop when I did postdoctoral training with the late Professor Minor J. (“Jud”) Coon on cytochrome P450 (P450) at the University of Michigan. I did not imagine that I would be working on the same enzymes for the rest of my career, but here I am. I was able to hold a good job and provide for my family, and I also came to learn a lot about the pharmaceutical enterprise, which is absolutely vital to health in our society. Along the way I was able to make a few research contributions that have had some bearing on how drugs are developed. Just as importantly, I trained a number of young people who went on to advance the field in industry, academia, and administration; four of them have also contributed Minireview articles in this issue. Overall, I have been able to do a lot of interesting things and to meet many good people. For all of this, I am very grateful.
Changes and Progress
Fifty years ago, there was a heavy focus on studying drug metabolism in vivo in experimental animals. Predicting drug metabolism and pharmacokinetics was slow and hard, and often the animal work did not extrapolate well to humans. As I have said sometimes, people were good at making drugs to cure rats. Poor pharmacokinetics in humans was a major source of attrition for drug candidates, if not the major one ( Prentis et al., 1988 ; Kola and Landis, 2004 ). Additionally, most drug doses were high, understandably, because in vitro pharmacology discovery programs were focused on getting IC 50 and inhibition constant (K i ) values in the micromolar range, not nanomolar.
What did we know at the molecular level? When I joined Jud Coon’s laboratory in 1973, no P450 had yet been purified to homogeneity. There was still debate about whether rats had one (hepatic) P450, or two, or more. For an interesting read, look at the published discussions following the talks at the first Microsomes and Drug Oxidations meeting in 1968. No one really knew much of anything about human P450s, except that they (or only a single one?) existed. There was no knowledge of even primary structures (sequences) of any P450s—recombinant DNA technology would not achieve this until 1982 ( Fujii-Kuriyama et al., 1982 ). P450 induction had been discovered by Remmer (1959 ) and James and Elizabeth Miller, with their graduate student Allan Conney ( Conney et al., 1956 ), but no one understood the mechanism nor was even sure if new protein synthesis was required.
Transporters were not even considered in the 1970s. As late as the early 1990s, the general dogma was that drugs are hydrophobic and enter cells by passive diffusion. Metabolites were usually more water-soluble than the parent drugs and therefore excreted, going back to some of the original concepts of R. T. Williams ( Williams, 1947 ). Although active transport was known, it was largely studied in bacteria and unrecognized regarding a role in drug metabolism. Although a phenomenon known as multiple drug resistance was known to be operative in cancer cells and relevant to therapy ( Endicott et al., 1987 ), the significance of these proteins in drug metabolism was not appreciated until later ( Schuetz et al., 1995 ). Today, transporters are an important aspect of drug metabolism.
When I started my own laboratory at Vanderbilt in 1975, we focused on “classical” enzyme purification as a means to characterizing P450s. In retrospect, this was not terribly “hypothesis driven” work, but it was important at the time and fortunately, I did get funded. Our first efforts were directed toward rat liver P450s, and eventually we purified nine of them ( Guengerich et al., 1982a ; Larrey et al., 1984 ). We were not alone in this effort; we had some serious and stimulating competition ( Ryan et al., 1975 ). With these P450s in hand, we could define catalytic specificity. In addition, we used antibodies (made in our own laboratory) and adapted newly discovered immunoelectrophoretic approaches ( Towbin et al., 1979 ) (later termed “Western blotting”) to quantify individual P450s and reach some new conclusions about P450 regulation ( Guengerich et al., 1982a ,b; Dannan et al., 1983 ).
We were finding significant differences between the rat and rabbit P450s, and the quest to understand the human liver P450s became very pressing. At first, we were just purifying red proteins from columns and trying to see what these P450s would do, candidly speaking ( Wang et al., 1980 ; Wang et al., 1983 ). We were finally able to obtain high quality human liver samples for our studies through some chance connections ( Mukhopadhyay, 2012 ).
I was impressed with the research that Robert Smith was doing on pharmacogenetics at what was then St. Mary’s Hospital Medical School in London. His work on the polymorphism of debrisoquine 4-hydroxylation showed that a single P450 gene locus could be very dominant in the metabolism of a drug, i.e., there could be considerable specificity of an individual P450 for a substrate ( Mahgoub et al., 1977 ; Tucker et al., 1977 ). Accordingly, we proceeded to use catalytic assays to monitor our purifications from liver. This approach was very technically challenging because of the presence of detergents and the difficulty of doing assays, such as debrisoquine hydroxylation (combined gas chromatography-mass spectrometry), but ultimately, we purified what are known today as P450s 1A2, 2A6, 2C8, 2C9, 2D6, and 3A4 from human liver ( Distlerath et al., 1985 ; Guengerich et al., 1986 ; Shimada et al., 1986 ; Yun et al., 1991 ).
With these P450s in hand, plus antibodies we raised to them, we were able to define the selectivity of these P450s toward drugs, some steroids, and then many chemical carcinogens ( Shimada et al., 1989 ). I suppose that all of this might have happened later anyway with recombinant DNA work and heterologous protein expression. By the same logic, one could dismiss Alexander Graham Bell’s invention of the telephone in that we no longer use it very much.
With this background and with much more work by many laboratories, particularly in the heterologous expression of P450s, it has been possible to learn much about 3-dimensional structures and mechanisms of induction and inhibition ( Fig. 1 ). The Human Genome Project finally answered the question of how many P450 genes people have (57). I consider it very fortuitous that a small set of human liver P450s—1A2, 2C9, 2D6, 3A4 (vide supra), and 2C19—catalyze ∼90% of the drug oxidation reactions ( Guengerich, 2015 ; Bhutani et al., 2021 ). Although this set does not work on all drugs, it could have been much more complicated if the situation were as complex as plant P450 biochemistry (>1,000 P450 genes in wheat, plus multiple reductases in plants).

Progress in drug metabolism. Dates are noted. (A) SDS—polyacrylamide gel electrophoresis (tube gels) of several rat P450s. Current names: lane 1, P450 2C11; lane 2, P450 2B1; lane 3, P450 2C6; lane 4, P450 2B2 ( Guengerich, 1977 ). (B) Structure of P450 3A4 (no ligand). Protein Data Bank 1TQN ( Yano et al., 2004 ). (C) Drug interaction table: https://drug-interactions.medicine.iu.edu/MainTable.aspx (accessed < June 11, 2022 >).
Where Are We Today?
The knowledge about P450 has been very useful in several regards. Whereas I have focused on P450s, similar progress has been made with other enzymes involved in drug metabolism and with transporters. Logical comparisons can be made between human and experimental animals, which are still necessary in the contexts of understanding pharmacological actions and safety assessment. In vitro results with human enzyme systems can be used reasonably well to model and predict human pharmacokinetics and drug-drug interactions. Pharmacokinetic changes caused by single-nucleotide variants (SNVs) can be identified. The improvements in analytical chemistry over the past 50 years have been amazing, both in terms of the inherent capability, sensitivity, and throughput (especially NMR and particularly combined liquid chromatography-mass spectrometry, which is only ∼35 years old). Our knowledge of chemical mechanisms of P450 reactions [particularly roles of Compound I (FeO 3+ )] has led to logical approaches to understanding metabolic pathways ( Guengerich, 2001 ; Guengerich and Yoshimoto, 2018 ). We know a considerable amount about the auxiliary flavoprotein NADPH-P450 reductase, which delivers the electrons to most of the P450s, and also about adrenodoxin and cytochrome b 5 (which can deliver electrons in some cases), in terms of the basic chemistry and also the relevance of genetic variations in these proteins and their relevance to disease states. Of the 57 human P450s, at least 25 have one or more X-ray crystal structures [plus at least two good animal orthologs in cases where a human structure is not yet available (i.e., 4B1, 24A1)]. Overall, drug metabolism has been a real success story in the application of biochemical approaches to practical problems of pharmaceutical development, and I am thankful to have had at least some role in this over the past 47+ years.
A Continued Need for Drug Metabolism
The above description all sounds good, so do we still need drug metabolism and pharmacokinetics departments in the pharmaceutical industry and biochemists and pharmacologists studying basic research in drug metabolism? The answers are both, YES!
Oral, low molecular weight drugs will continue to be important. We have seen this recently with the coronavirus disease 2019 therapies. Today, many drugs are very potent (inhibition constant values in the low nanomolar range), practical, and are available at overall low cost (despite the naysayers, they actually have short patent lives on the market). Another reason for good drug metabolism science is that the regulatory expectations have become higher, e.g., US Food and Drug Administration, European Medicines Agency, Japanese Pharmaceuticals and Medical Devices Agency. With advances in analytical chemistry has come the expectation to define even more minor metabolites, and there are US Food and Drug Administration Metabolites in Safety Testing regulations regarding “disproportionate [human] metabolites” in species comparisons ( Schadt et al., 2018 ). Time-dependent inhibition is still a problem, particularly with P450 3A4 (Eng et al ., 2020 ). Related to this are drug-drug interactions, often with P450 3A4 and P-glycoprotein and some other transporters, which are still a problem, even fatal ( Yu et al., 2018 ). Toxicology (safety assessment) problems are a major cause of attrition of drug candidates ( Kola and Landis, 2004 ), particularly hepatic and cardiovascular issues. In particular, drug-induced liver injury is still difficult to predict and often involves drug metabolism.
New Challenges and Opportunities
With every problem or challenge, there is an opportunity to make an important contribution. At the outset, I raise the caveat that the following are some of my ideas but that another person may well have a different list. Regardless, I seriously doubt that I will be able to solve all of these challenges myself in the time I have left to play the game. The future of drug metabolism is still bright for young scientists looking for meaningful careers.
Analytical chemistry will get even better. I am impressed with every new combined liquid chromatography-mass spectrometry model that comes out (but do not have the resources to buy). Even this may change though, at least the liquid chromatography component. UPLC can be fast but still takes time. Direct injection high resolution mass spectrometry approaches are already being used in metabolomics research ( Sarvin et al., 2020 ). On the horizon is acoustic ejection mass spectrometry for very high throughput analysis, which has serious potential ( Simon et al., 2021 ; Zhang et al., 2021 )—3600 samples/h with 0.01 µ l per shot! [In the P450 3A4 purification work ( Guengerich et al., 1986 ) I thought I was doing well manually injecting one sample from a nifedipine oxidation reaction with a (P450) column fraction myself every 3 minutes, using the “new” 6.2 mm × 80 mm Zorbax columns and running at a flow rate of 4 ml min −1 .] Another technique with potential is crystalline sponge X-ray diffraction analysis ( Rosenberger et al., 2020 ; Rosenberger et al., 2021 ). A porous metal coordination complex functions as a host crystal, and µ g quantities of “guest” ligands can be added and induced to crystallize. The method has the power to determine 3-dimensional structures of drug metabolites with as little as 5 µ g, surpassing 2-D NMR in sensitivity and time for detailed analysis ( Rosenberger et al., 2021 ), although only ∼2/3 of the molecules tested are successful to date.
Another developing area is artificial intelligence (AI), which has already been employed extensively in predictions of genetic toxicology ( Cunningham et al., 2004 ) and in regioselectivity of drug metabolism ( de Bruyn Kops et al., 2021 ). Most of the logic is based on literature precedents and AI, not on inherent structural docking and other physical principles. Although some of the algorithms have achieved impressive results (particularly in picking the top three “hot spots”), there are still many exceptions [e.g., hydroxylations of the angular methyl groups (C18, C19) of 4,5-dihydrotestosterone by P450 3A4 ( Cheng et al., 2012 )]. AI predictions of rates of metabolism are even more challenging, aside from Hammett series of molecules ( Burka et al., 1985 ). Although, with enough dataset entries, even this might be possible someday.
Another area of potential is toxicity and safety assessment. Even predicting drug-drug interactions is difficult ( Eng et al., 2020 ). These can be quite variable depending on the perpetrator and victim pair. Prediction of toxicities such as drug-induced liver injury is even more challenging because of the biologic complexity of tissues, even with in vitro data, but some progress is being made with AI approaches ( Li et al., 2021 ). Predicting toxicities in other organs is also difficult, but some advances have been made in the context of biomarkers for both animals and humans ( Harrill et al., 2009 ; Vazquez et al., 2020 ).
The field of pharmacogenetics developed with some of the enzymes in drug metabolism ( Motulsky, 1957 ; Kalow, 1962 ) but accelerated with the work of Smith and others on what is now P450 2D6 ( Mahgoub et al., 1977 ; Tucker et al., 1977 ; Caldwell, 2006 ). The input of molecular biology also changed the landscape ( Nebert et al., 1981 ; Fujii-Kuriyama et al., 1982 ; Gonzalez et al., 1988 ). Although the early pharmacogenetic studies were interpreted in the context of “extensive” and “poor” metabolizers (fast and slow) ( Mahgoub et al., 1977 ), today we know that there are hundreds of SNVs with at least some of the P450s ( https://www.pharmvar.org/gene/CYP2D6 ), and bimodal interpretation of the results is much too simplistic. We also know that >100 clinically relevant SNVs of the steroid 21-hydroxylase P450 21A2 exist (grouped into three phenotypic categories) ( Wang et al., 2017 ). Understanding the links between single amino acid structural changes and function in these (and other) enzymes is challenging, and few natural P450 variants have been crystallized ( Parikh et al., 2020 ). The root of the problem is seen in the application of the Eyring equation
(where R is the universal gas constant, T is the absolute temperature, N A is Avagadro’s number, and h is Planck’s constant), in that a 10-fold variation in enzyme activity ( k obs ) is linked with a free energy change (Δ G ) of 1.3 kcal mol −1 , less than a single hydrogen bond ( Wang et al., 2017 ). Can we ever understand or predict rates of drug metabolism of variants? Also, realize that different coding region SNVs can yield different changes with different drug oxidation reactions ( Takanashi et al., 2000 ). At a clinical level, there is still limited use of what we do know about phenotypes of SNVs, with the most prominent examples being thiopurine S -methylation ( Lennard, 2014 ), warfarin/P450 2C9 ( Higashi et al., 2002 ), and clopidogrel/P450 2C19 (Pare et al., 2010). Some use of P450 2D6 SNVs has been made with iloperidone (FANAPT) ( https://fanaptpro.com/wp-content/uploads/2015/02/Fanapt-Prescribing-Information.pdf ) and possibly other neurological drugs ( Haslemo et al., 2019 ). The SNVs in steroid-metabolizing P450s have more dramatic clinical consequences ( Miller and Auchus, 2011 ), but the effects of new SNVs are still rather unpredictable. For instance, development of drugs that can rescue poor phenotypes of the steroid 21-hydroxylase P450 21A2 is a formidable challenge.
Although some functions are now associated with most of the human P450s, there are a few recalcitrant “orphans” ( Guengerich, 2015 ), e.g., 4X1 and 20A1. Beyond this, there is still a concern about what the relatively slow rate of fatty acid oxidations catalyzed by some of the P450s really mean ( Stark et al., 2008 ; Fekry et al., 2019 ). Are we missing important roles? Are the functions of the xenobiotic-metabolizing P450s only general cellular protection (e.g., against ingested natural products) or do more of these P450s have physiologically relevant substrates?
Of the 57 human P450s, 50 are inherently microsomal and seven are mitochondrial ( Guengerich, 2015 ). Most of the mitochondrial P450s seem to all be essential (27C1?) and have important physiologic substrates, but we also know that at least some of these can have roles in drug metabolism and even bioactivation ( Zhang et al., 2012 ; Rendic and Guengerich, 2018 ). Are there more drugs oxidized in mitochondria? Narayan Avadhani and his associates have shown that some of the microsomal P450s can also be modified and relocate to the mitochondria ( Avadhani et al., 2011 ). They appear to then use adrenodoxin as a source of electrons for reduction, but we know little about the details of these interactions. There are still major questions about the abundance of the mitochondrial P450s and their accessory proteins in the relevant zones of the extrahepatic tissues where they are found.
Although we are developing concepts of how P450s interact with NADPH-P450 reductase ( Cheng et al., 2021 ) and the relevance of genetic variations in these proteins and their relevance to disease states is recognized ( Riddick et al., 2013 ; Burkhard et al., 2017 ), there are other interactions that remain poorly understood. The interaction of adrenodoxin has only been studied with some of the mitochondrial P450s ( Lambeth and Kriengsiri, 1985 ; Beilke et al., 2002 ; Brixius-Anderko and Scott, 2021 ; Glass et al., 2021 ). Although it has been >50 years since the discovery of a role for cytochrome b 5 in a P450 reaction ( Hildebrandt and Estabrook, 1971 ; Guengerich, 2022 ), there are still major questions about mechanisms and relevance in vivo ( McLaughlin et al., 2010 ). b 5 may stimulate or inhibit P450 reactions and is most essential in the P450 17A1 lyase reaction ( Katagiri et al., 1995 ). Stimulation may or may not involve electron transfer with individual P450s ( Yamazaki et al., 2002 ). Recently retinoid-binding proteins have been shown to deliver retinoids directly to P450 Family 26 enzymes ( Zhong et al., 2018 ) and P450 27C1 ( Glass and Guengerich, 2021 ), although structural details are unknown. The demonstrated roles of binding proteins of the retinoid-metabolizing P450s raise the question of whether fatty acid binding proteins may also be involved in substrate delivery. There is still a glaring need for more structural work on binary complexes of P450s with accessory proteins.
The clinical relevance of P450s has been clearly demonstrated in drug-drug interactions and endocrinology, but the situation is less clear in other areas. Chemical carcinogenesis was one of the areas that fueled much of the early research with P450s and several other drug-metabolizing enzymes. Although roles for many of these enzymes have been clearly implicated in animal models ( Guengerich, 1988 ), the situation is less clear in humans and studies of SNVs have not been very definitive. Unfortunately, interest in this area has waned, certainly with the funders at the National Institutes of Health and much of the cancer research community. Other areas that have shown some potential relevance of P450s and SNVs include hypertension ( Gainer et al., 2005 ) and neurologic disease ( Cheng et al., 2013 ).
P450s can also be drug targets, e.g., P450s 5A1, 11B2, 17A1, 19A1. Inhibition of P450 19A1 steroid aromatase activity is an established means of treating breast and other estrogen-dependent cancers. Some other P450s are drug targets and have drugs to inhibit them, but these could be improved, e.g., P450 17A1 ( Bird and Abbott, 2016 ). More P450s, including those essential in normal settings, are potential drug targets. Finally, inhibiting P450s of parasites and other infectious agents is an established approach to treating very important disease problems, e.g., fungal infections, and frequent targets are P450 51 enzymes ( Friggeri et al., 2014 ). P450s have also been considered as targets in treating tuberculosis ( McLean et al., 2007 ).
Other Issues Related To Drug Metabolism
There are several “nonscience” issues of concern in the area of drug metabolism, although they really do involve science.
First, the past two years have shown us how fragile our interactive networks can be. There are new approaches to communication, but the live meetings have been sorely missed. Some of the important things I have to look back on in my career are the lessons I learned in my travels and the people I have met. We had to postpone the biennial International Conference on Cytochrome P450 (ICCP450) from 2021 to 2022, which has now occurred by the time this review is published. I worry about both diseases and international politics disrupting future meetings, not only ICCP450 but also others.
Scientific publishing has changed considerably in recent years. The challenges have been great for historic society-based journals (e.g., Drug Metabolism and Disposition , The Journal of Biologic Chemistry ). Although the development of electronic capability has been very useful, economic pressures have been an issue. Will journals edited by working scientists survive? Will books exist or have they become dinosaurs (the offices of our junior faculty certainly do not resemble mine!)? I have serious concerns about the development of nonreviewed publications (which will remain nameless here). Exactly what is scientific communication going to look like in 50 more years?
Finally, who will be training the next generation of drug metabolism scientists? I see contemporaries disappearing but limited enthusiasm of universities for hiring in the area. Would I even be hired today? What do we need to be teaching graduate students and postdocs to prepare them for the pharmaceutical industry and other careers? What new subject areas need to be added to the repertoire of a trainee—and which basic ones need to be retained? Who will be the faculty doing this and, most importantly, will the National Institutes of Health and other agencies fund individuals to maintain a viable cadre in this field?
Conclusions
The science of drug metabolism really began in the 19 th Century ( Caldwell, 2006 ; Guengerich, 2018 ) (see also https://www.issx.org/page/History ), but the bulk of what we know has been learned in the last 50 years. I have been privileged to have had a role in this. Drug metabolism has been a classic success in the application of basic science to important health problems. For reasons described, drug metabolism will continue to play a vital role in the process of discovering and developing drugs. A number of opportunities exist for further development. There are scientific and other challenges ahead, as there were in the past.
Acknowledgments
I thank K. Trisler for her assistance in the preparation of the manuscript and the editors for the opportunity to contribute this Minireview. Finally, I thank the 22 graduate students, 141 postdocs and visiting scientists, and all others who have worked in this laboratory and contributed, as well as our collaborators. Their individual efforts have made our research possible but are too many to mention in the available space.
Finally, I would like to dedicate this article to the memory of Prof. Michael R. Waterman, my friend and colleague for over 20 years. Mike died in November 2021 during the start of this draft. He made many contributions to the field of P450 research and is missed by all who knew him.
Abbreviations
artificial intelligence
cytochrome P450
single-nucleotide variation
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Guengerich.
This work was supported in part by the National Institute of General Medical Sciences (Grant R01-GM118122) (to F.P.G.).
dx.doi.org/10.1124/dmd.121.000739 .
- Gillette JR, Conney AH, Cosmides GJ, Estabrook RW, Fouts JR, Mannering GJ, eds (1968) in Microsomes and Drug Oxidations, pp 3–547, Academic Press, Bethesda, MD. [ Google Scholar ]
- Avadhani NG, Sangar MC, Bansal S, Bajpai P (2011) Bimodal targeting of cytochrome P450s to endoplasmic reticulum and mitochondria: the concept of chimeric signals. FEBS J 278:4218–4229. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Beilke D, Weiss R, Löhr F, Pristovsek P, Hannemann F, Bernhardt R, Rüterjans H (2002) A new electron transport mechanism in mitochondrial steroid hydroxylase systems based on structural changes upon the reduction of adrenodoxin. Biochemistry 41:7969–7978. [ DOI ] [ PubMed ] [ Google Scholar ]
- Bhutani P, Joshi G, Raja N, Bachhav N, Rajanna PK, Bhutani H, Paul AT, Kumar R (2021) US FDA approved drugs from 2015-June 2020: a perspective. J Med Chem 64:2339–2381. [ DOI ] [ PubMed ] [ Google Scholar ]
- Bird IM, Abbott DH (2016) The hunt for a selective 17,20 lyase inhibitor; learning lessons from nature. J Steroid Biochem Mol Biol 163:136–146. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Brixius-Anderko S, Scott EE (2021) Structural and functional insights into aldosterone synthase interaction with its redox partner protein adrenodoxin. J Biol Chem 296:100794. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Burka LT, Guengerich FP, Willard RJ, Macdonald TL (1985) Mechanism of cytochrome P-450 catalysis. mechanism of N -dealkylation and amine oxide deoxygenation. J Am Chem Soc 107:2549–2551. [ Google Scholar ]
- Burkhard FZ, Parween S, Udhane SS, Flück CE, Pandey AV (2017) P450 Oxidoreductase deficiency: analysis of mutations and polymorphisms. J Steroid Biochem Mol Biol 165:38–50. [ DOI ] [ PubMed ] [ Google Scholar ]
- Caldwell J (2006) Drug metabolism and pharmacogenetics: the British contribution to fields of international significance. Br J Pharmacol 147:S89–S99. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Cheng J, Zhen Y, Miksys S, Beyoğlu D, Krausz KW, Tyndale RF, Yu A, Idle JR, Gonzalez FJ (2013) Potential role of CYP2D6 in the central nervous system. Xenobiotica 43:973–984. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Cheng Q, Sohl CD, Yoshimoto FK, Guengerich FP (2012) Oxidation of dihydrotestosterone by human cytochromes P450 19A1 and 3A4. J Biol Chem 287:29554–29567. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Cheng S, Bo Z, Hollenberg P, Osawa Y, Zhang H (2021) Amphipol-facilitated elucidation of the functional tetrameric complex of full-length cytochrome P450 CYP2B4 and NADPH-cytochrome P450 oxidoreductase. J Biol Chem 296:100645. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Conney AH, Miller EC, Miller JA (1956) The metabolism of methylated aminoazo dyes. V. evidence for induction of enzyme synthesis in the rat by 3-methylcholanthrene. Cancer Res 16:450–459. [ PubMed ] [ Google Scholar ]
- Cunningham AR, Cunningham SL, Rosenkranz HS (2004) Structure-activity approach to the identification of environmental estrogens: the MCASE approach. SAR QSAR Environ Res 15:55–67. [ DOI ] [ PubMed ] [ Google Scholar ]
- Dannan GA, Guengerich FP, Kaminsky LS, Aust SD (1983) Regulation of cytochrome P-450. Immunochemical quantitation of eight isozymes in liver microsomes of rats treated with polybrominated biphenyl congeners. J Biol Chem 258:1282–1288. [ PubMed ] [ Google Scholar ]
- de Bruyn Kops C, Šícho M, Mazzolari A, Kirchmair J (2021) GLORYx: prediction of the metabolites resulting from phase 1 and phase 2 biotransformations of xenobiotics. Chem Res Toxicol 34:286–299. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Distlerath LM, Reilly PE, Martin MV, Davis GG, Wilkinson GR, Guengerich FP (1985) Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O -deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem 260:9057–9067. [ PubMed ] [ Google Scholar ]
- Endicott JA, Juranka PF, Sarangi F, Gerlach JH, Deuchars KL, Ling V (1987) Simultaneous expression of two P-glycoprotein genes in drug-sensitive Chinese hamster ovary cells. Mol Cell Biol 7:4075–4081. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Eng H, Tseng E, Cerny MA, Goosen TC, Obach RS (2020) Cytochrome P450 3A time-dependent inhibition assays are too sensitive for identification of drugs causing clinically significant drug-drug interactions: a comparison of human liver microsomes and hepatocytes and definition of boundaries for inactivation rate constants. Drug Metab Dispos 49:442–450. [ DOI ] [ PubMed ] [ Google Scholar ]
- Fekry MI, Xiao Y, Berg JZ, Guengerich FP (2019) A role for the orphan human cytochrome P450 2S1 in polyunsaturated fatty acid ω-1 hydroxylation using an untargeted metabolomic approach. Drug Metab Dispos 47:1325–1332. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Friggeri L, Hargrove TY, Rachakonda G, Williams AD, Wawrzak Z, Di Santo R, De Vita D, Waterman MR, Tortorella S, Villalta F, et al. (2014) Structural basis for rational design of inhibitors targeting Trypanosoma cruzi sterol 14α-demethylase: two regions of the enzyme molecule potentiate its inhibition. J Med Chem 57:6704–6717. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Fujii-Kuriyama Y, Mizukami Y, Kawajiri K, Sogawa K, Muramatsu M (1982) Primary structure of a cytochrome P-450: coding nucleotide sequence of phenobarbital-inducible cytochrome P-450 cDNA from rat liver. Proc Natl Acad Sci USA 79:2793–2797. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Gainer JV, Bellamine A, Dawson EP, Womble KE, Grant SW, Wang Y, Cupples LA, Guo CY, Demissie S, O’Donnell CJ, et al. (2005) Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation 111:63–69. [ DOI ] [ PubMed ] [ Google Scholar ]
- Glass SM, Guengerich FP (2021) Cellular retinoid-binding proteins transfer retinoids to human cytochrome P450 27C1 for desaturation. J Biol Chem 297:101142. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Glass SM, Webb SN, Guengerich FP (2021) Binding of cytochrome P450 27C1, a retinoid desaturase, to its accessory protein adrenodoxin. Arch Biochem Biophys 714:109076. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Gonzalez FJ, Skoda RC, Kimura S, Umeno M, Zanger UM, Nebert DW, Gelboin HV, Hardwick JP, Meyer UA (1988) Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature 331:442–446. [ DOI ] [ PubMed ] [ Google Scholar ]
- Guengerich FP (1977) Separation and purification of multiple forms of microsomal cytochrome P-450. Activities of different forms of cytochrome P-450 towards several compounds of environmental interest. J Biol Chem 252:3970–3979. [ PubMed ] [ Google Scholar ]
- Guengerich FP (1988) Roles of cytochrome P-450 enzymes in chemical carcinogenesis and cancer chemotherapy. Cancer Res 48:2946–2954. [ PubMed ] [ Google Scholar ]
- Guengerich FP (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 14:611–650. [ DOI ] [ PubMed ] [ Google Scholar ]
- Guengerich FP(2015) Human cytochrome P450 enzymes, in: Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano PR, ed), pp 523–785, Springer, New York. [ Google Scholar ]
- Guengerich FP (2018) Introduction and historical perspective, in Biotransformation (Guengerich FP, ed) pp 1–7, Elsevier, Oxford, UK. [ Google Scholar ]
- Guengerich FP (2022) On ‘Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions’ by Alfred Hildebrandt and Ronald W. Estabrook Arch Biochem Biophys 726:109177. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Guengerich FP, Dannan GA, Wright ST, Martin MV, Kaminsky LS (1982a) Purification and characterization of liver microsomal cytochromes P-450: electrophoretic, spectral, catalytic, and immunochemical properties and inducibility of eight isozymes isolated from rats treated with phenobarbital or β -naphthoflavone. Biochemistry 21:6019–6030. [ DOI ] [ PubMed ] [ Google Scholar ]
- Guengerich FP, Martin MV, Beaune PH, Kremers P, Wolff T, Waxman DJ (1986) Characterization of rat and human liver microsomal cytochrome P-450 forms involved in nifedipine oxidation, a prototype for genetic polymorphism in oxidative drug metabolism. J Biol Chem 261:5051–5060. [ PubMed ] [ Google Scholar ]
- Guengerich FP, Wang P, Davidson NK (1982b) Estimation of isozymes of microsomal cytochrome P-450 in rats, rabbits, and humans using immunochemical staining coupled with sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Biochemistry 21:1698–1706. [ DOI ] [ PubMed ] [ Google Scholar ]
- Guengerich FP, Yoshimoto FK (2018) Formation and cleavage of C-C bonds by enzymatic oxidation-reduction reactions. Chem Rev 118:6573–6655. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Harrill AH, Ross PK, Gatti DM, Threadgill DW, Rusyn I (2009) Population-based discovery of toxicogenomics biomarkers for hepatotoxicity using a laboratory strain diversity panel. Toxicol Sci 110:235–243. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Haslemo T, Eliasson E, Jukić MM, Ingelman-Sundberg M, Molden E (2019) Significantly lower CYP2D6 metabolism measured as the O/N-desmethylvenlafaxine metabolic ratio in carriers of CYP2D6*41 versus CYP2D6*9 or CYP2D6*10: a study on therapeutic drug monitoring data from 1003 genotyped Scandinavian patients. Br J Clin Pharmacol 85:194–201. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Higashi MK, Veenstra DL, Kondo LM, Wittkowsky AK, Srinouanprachanh SL, Farin FM, Rettie AE (2002) Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 287:1690–1698. [ DOI ] [ PubMed ] [ Google Scholar ]
- Hildebrandt A, Estabrook RW (1971) Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Arch Biochem Biophys 143:66–79. [ DOI ] [ PubMed ] [ Google Scholar ]
- Kalow W (1962) Pharmacogenetics, W. B. Saunders, Philadelphia. [ Google Scholar ]
- Katagiri M, Kagawa N, Waterman MR (1995) The role of cytochrome b 5 in the biosynthesis of androgens by human P450c17. Arch Biochem Biophys 317:343–347. [ DOI ] [ PubMed ] [ Google Scholar ]
- Kola I, Landis J (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3:711–715. [ DOI ] [ PubMed ] [ Google Scholar ]
- Lambeth JD, Kriengsiri S (1985) Cytochrome P-450scc-adrenodoxin interactions. Ionic effects on binding, and regulation of cytochrome reduction by bound steroid substrates. J Biol Chem 260:8810–8816. [ PubMed ] [ Google Scholar ]
- Larrey D, Distlerath LM, Dannan GA, Wilkinson GR, Guengerich FP (1984) Purification and characterization of the rat liver microsomal cytochrome P-450 involved in the 4-hydroxylation of debrisoquine, a prototype for genetic variation in oxidative drug metabolism. Biochemistry 23:2787–2795. [ DOI ] [ PubMed ] [ Google Scholar ]
- Lennard L (2014) Implementation of TPMT testing. Br J Clin Pharmacol 77:704–714. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Li T, Tong W, Roberts R, Liu Z, Thakkar S (2021) DeepDILI: deep learning-powered drug-induced liver injury prediction using model-level representation. Chem Res Toxicol 34:550–565. [ DOI ] [ PubMed ] [ Google Scholar ]
- Mahgoub A, Idle JR, Dring LG, Lancaster R, Smith RL (1977) Polymorphic hydroxylation of debrisoquine in man. Lancet 2:584–586. [ DOI ] [ PubMed ] [ Google Scholar ]
- McLaughlin LA, Ronseaux S, Finn RD, Henderson CJ, Wolf CR (2010) Deletion of microsomal cytochrome b 5 profoundly affects hepatic and extrahepatic drug metabolism. Mol Pharmacol 78:269–278. [ DOI ] [ PubMed ] [ Google Scholar ]
- McLean KJ, Dunford AJ, Neeli R, Driscoll MD, Munro AW (2007) Structure, function and drug targeting in Mycobacterium tuberculosis cytochrome P450 systems. Arch Biochem Biophys 464:228–240. [ DOI ] [ PubMed ] [ Google Scholar ]
- Miller WL, Auchus RJ (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 32:81–151. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Motulsky AG (1957) Drug reactions enzymes, and biochemical genetics. J Am Med Assoc 165:835–837. [ DOI ] [ PubMed ] [ Google Scholar ]
- Mukhopadhyay R (2012) Human cytochrome P450s: the work of Frederick Peter Guengerich. J Biol Chem 287:15798–15800. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Nebert DW, Eisen HJ, Negishi M, Lang MA, Hjelmeland LM, Okey AB (1981) Genetic mechanisms controlling the induction of polysubstrate monooxygenase (P-450) activities. Annu Rev Pharmacol Toxicol 21:431–462. [ DOI ] [ PubMed ] [ Google Scholar ]
- Paré G, Mehta SR, Yusuf S, Anand SS, Connolly SJ, Hirsh J, Simonsen K, Bhatt DL, Fox KA, Eikelboom JW (2010) Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med 363:1704–1714. [ DOI ] [ PubMed ] [ Google Scholar ]
- Parikh SJ, Evans CM, Obi JO, Zhang Q, Maekawa K, Glass KC, Shah MB (2020) Structure of cytochrome P450 2C9*2 in complex with an anti-hypertensive drug losartan: Insights into the effect of genetic polymorphism. Mol Pharmacol 98:529–539. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Prentis RA, Lis Y, Walker SR (1988) Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964-1985). Br J Clin Pharmacol 25:387–396. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Remmer H (1959) [The acceleration of evipan oxidation and the demethylation of methylaminopyrine by barbiturates]. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol 237:296–307. [ PubMed ] [ Google Scholar ]
- Rendić SP, Peter Guengerich F (2018) Human cytochrome P450 enzymes 5-51 as targets of drugs and natural and environmental compounds: mechanisms, induction, and inhibition - toxic effects and benefits. Drug Metab Rev 50:256–342. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Riddick DS, Ding X, Wolf CR, Porter TD, Pandey AV, Zhang Q-Y, Gu J, Finn RD, Ronseaux S, McLaughlin LA, et al. (2013) NADPH-cytochrome P450 oxidoreductase: roles in physiology, pharmacology, and toxicology. Drug Metab Dispos 41:12–23. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Rosenberger L, von Essen C, Khutia A, Kühn C, Georgi K, Hirsch AKH, Hartmann RW, Badolo L (2021) Crystalline sponge affinity screening: a fast tool for soaking condition optimization without the need of X-ray diffraction analysis. Eur J Pharm Sci 164:105884. [ DOI ] [ PubMed ] [ Google Scholar ]
- Rosenberger L, von Essen C, Khutia A, Kühn C, Urbahns K, Georgi K, Hartmann RW, Badolo L (2020) Crystalline sponges as a sensitive and fast method for metabolite identification: application to gemfibrozil and its phase I and II metabolites. Drug Metab Dispos 48:587–593. [ DOI ] [ PubMed ] [ Google Scholar ]
- Ryan D, Lu AYH, West S, Levin W (1975) Multiple forms of cytochrome P-450 in phenobarbital- and 3-methylcholanthrene-treated rats. Separation and spectral properties. J Biol Chem 250:2157–2163. [ PubMed ] [ Google Scholar ]
- Sarvin B, Lagziel S, Sarvin N, Mukha D, Kumar P, Aizenshtein E, Shlomi T (2020) Fast and sensitive flow-injection mass spectrometry metabolomics by analyzing sample-specific ion distributions. Nat Commun 11:3186. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Schadt SBister BChowdhury SKFunk CHop CECAHumphreys WGIgarashi FJames ADKagan MKhojasteh SC, et al. (2018) A decade in the MIST: learnings from investigations of drug metabolites in drug development under the “Metabolites in Safety Testing” regulatory guidance. Drug Metab Dispos 46:865–878. [ DOI ] [ PubMed ] [ Google Scholar ]
- Schuetz EG, Furuya KN, Schuetz JD (1995) Interindividual variation in expression of P-glycoprotein in normal human liver and secondary hepatic neoplasms. J Pharmacol Exp Ther 275:1011–1018. [ PubMed ] [ Google Scholar ]
- Shimada T, Iwasaki M, Martin MV, Guengerich FP (1989) Human liver microsomal cytochrome P-450 enzymes involved in the bioactivation of procarcinogens detected by umu gene response in Salmonella typhimurium TA 1535/pSK1002. Cancer Res 49:3218–3228. [ PubMed ] [ Google Scholar ]
- Shimada T, Misono KS, Guengerich FP (1986) Human liver microsomal cytochrome P-450 mephenytoin 4-hydroxylase, a prototype of genetic polymorphism in oxidative drug metabolism. Purification and characterization of two similar forms involved in the reaction. J Biol Chem 261:909–921. [ PubMed ] [ Google Scholar ]
- Simon RPHäbe TTRies RWinter MWang YFernández-Montalván ABischoff DRunge FReindl WLuippold AH, et al. (2021) Acoustic ejection mass spectrometry: a fully automatable technology for high-throughput screening in drug discovery. SLAS Discov 26:961–973. [ DOI ] [ PubMed ] [ Google Scholar ]
- Stark K, Dostalek M, Guengerich FP (2008) Expression and purification of orphan cytochrome P450 4X1 and oxidation of anandamide. FEBS J 275:3706–3717. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Takanashi K, Tainaka H, Kobayashi K, Yasumori T, Hosakawa M, Chiba K (2000) CYP2C9 Ile 359 and Leu 359 variants: enzyme kinetic study with seven substrates. Pharmacogenetics 10:95–104. [ DOI ] [ PubMed ] [ Google Scholar ]
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Tucker GT, Silas JH, Iyun AO, Lennard MS, Smith AJ (1977) Polymorphic hydroxylation of debrisoquine. Lancet 2:718. [ DOI ] [ PubMed ] [ Google Scholar ]
- Vazquez JH, Clemens MM, Allard FD, Yee EU, Kennon-McGill S, Mackintosh SG, Jaeschke H, Hambuchen MD, McGill MR (2020) Identification of serum biomarkers to distinguish hazardous and benign aminotransferase elevations. Toxicol Sci 173:244–254. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Wang C, Pallan PS, Zhang W, Lei L, Yoshimoto FK, Waterman MR, Egli M, Guengerich FP (2017) Functional analysis of human cytochrome P450 21A2 variants involved in congenital adrenal hyperplasia. J Biol Chem 292:10767–10778. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Wang P, Mason PS, Guengerich FP (1980) Purification of human liver cytochrome P-450 and comparison to the enzyme isolated from rat liver. Arch Biochem Biophys 199:206–219. [ DOI ] [ PubMed ] [ Google Scholar ]
- Wang PP, Beaune P, Kaminsky LS, Dannan GA, Kadlubar FF, Larrey D, Guengerich FP (1983) Purification and characterization of six cytochrome P-450 isozymes from human liver microsomes. Biochemistry 22:5375–5383. [ DOI ] [ PubMed ] [ Google Scholar ]
- Williams RT (1947) Detoxication Mechanisms, Wiley, New York. [ Google Scholar ]
- Yamazaki H, Nakamura M, Komatsu T, Ohyama K, Hatanaka N, Asahi S, Shimada N, Guengerich FP, Shimada T, Nakajima M, et al. (2002) Roles of NADPH-P450 reductase and apo- and holo-cytochrome b 5 on xenobiotic oxidations catalyzed by 12 recombinant human cytochrome P450s expressed in membranes of Escherichia coli . Protein Expr Purif 24:329–337. [ DOI ] [ PubMed ] [ Google Scholar ]
- Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF (2004) The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem 279:38091–38094. [ DOI ] [ PubMed ] [ Google Scholar ]
- Yu J, Zhou Z, Tay-Sontheimer J, Levy RH, Ragueneau-Majlessi I (2018) Risk of clinically relevant pharmacokinetic-based drug-drug interactions with drugs approved by the US Food and Drug Administration between 2013 and 2016. Drug Metab Dispos 46:835–845. [ DOI ] [ PubMed ] [ Google Scholar ]
- Yun CH, Shimada T, Guengerich FP (1991) Purification and characterization of human liver microsomal cytochrome P-450 2A6. Mol Pharmacol 40:679–685. [ PubMed ] [ Google Scholar ]
- Zhang DFlint OWang LGupta AWesthouse RAZhao WRaghavan NCaceres-Cortes JMarathe PShen G, et al. (2012) Cytochrome P450 11A1 bioactivation of a kinase inhibitor in rats: use of radioprofiling, modulation of metabolism, and adrenocortical cell lines to evaluate adrenal toxicity. Chem Res Toxicol 25:556–571. [ DOI ] [ PubMed ] [ Google Scholar ]
- Zhang HLiu CHua WGhislain LPLiu JAschenbrenner LNoell SDirico KJLanyon LFSteppan CM, et al. (2021) Acoustic ejection mass spectrometry for high-throughput analysis. Anal Chem 93:10850–10861. [ DOI ] [ PubMed ] [ Google Scholar ]
- Zhong G, Ortiz D, Zelter A, Nath A, Isoherranen N (2018) CYP26C1 is a hydroxylase of multiple active retinoids and interacts with cellular retinoic acid binding proteins. Mol Pharmacol 93:489–503. [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- View on publisher site
- PDF (966.7 KB)
- Collections
Similar articles
Cited by other articles, links to ncbi databases.
- Download .nbib .nbib
- Format: AMA APA MLA NLM
Add to Collections
IMAGES
VIDEO
COMMENTS
Drug metabolism/ Drug-drug interaction. Drug Metabolism: Mainly occurs in liver, but can occur in other locations like the lungs, kidneys, GIT, etc. 2 types of metabolism: 1) First Pass 2) Second Pass Aim of metabolism is to make drug more hydrophilic (water soluble) because water soluble drugs are more easily excreted in the kidneys.
Drug metabolism is a vital process that influences the fate of medications within the human body. It involves the enzymatic transformation of drugs into metabolites, which are more easily eliminated from the body. This process primarily occurs in the liver but also involves other organs. Drug metabolism serves various purposes, including drug
Drug metabolism essay - Grade: 88 drug collaboration drug metabolism: fundamentally happens in liver, yet can happen in different areas like the lungs, kidneys Skip to document University
Most drugs are xenobiotics, ie, chemical substances not naturally produced by the body. Xenobiotics undergo various body processes for detoxification, thus reducing their toxicity and allowing them to be readily available for excretion. These processes allow for the chemical modification of drugs into their metabolites and are known as drug metabolism or metabolic biotransformation.[1][2]
How does age affect drug metabolism: There are many physiological changes that occur with ageing. The changes have the potential to affect both drug disposition and metabolism. Drug metabolism is mainly functioned by the liver, its size, blood perfusion and synthetic capacity for proteins which all determine the rate of hepatic drug elimination[5].
Drug metabolism involves a series of biological and chemical processes through that endogenous substances are converted into more water-soluble substituents resultantly excreted out from the body. Drug metabolic pathways accomplished by phase I and phase II biotransformation reactions and various in vitro and in vivo models are employed for the ...
In such cases, called first-order elimination (or kinetics), the metabolism rate of the drug is a constant fraction of the drug remaining in the body (ie, the drug has a specific half-life). For example, if 500 mg is present in the body at time zero, after metabolism, 250 mg may be present at 1 hour and 125 mg at 2 hours (illustrating a half ...
5.4 Drug Metabolism Exploits Ubiquitous Themes in Chemistry. One of the remarkable features of hepatocytes and drug metabolism within the body is that there is a high efficiency and capacity to metabolize foreign molecules with a wide range of chemical diversity. Instead of developing a tailored nucleophile to react with specific drug, general ...
The primary objective of drug metabolism is to facilitate a drug's excretion by increasing its water solubility (hydrophilicity). The involved chemical modifications incidentally decrease or increase a drug's pharmacological activity and/or half-life, the most extreme example being the metabolic activation of inactive prodrugs into active drugs, e.g. of codeine into morphine by CYP2D6.
The systematic study of drug metabolism began in the 19th Century, but most of what we know now has been learned in the last 50 years. Drug metabolism continues to play a critical role in pharmaceutical development and clinical practice, as well as contributing to toxicology, chemical carcinogenesis, endocrinology, and drug abuse.